Zero-point renormalization of the band gap and temperature-dependent band gaps¶
This tutorial explains how to compute the electron self-energy due to phonons, obtain the zero-point renormalization (ZPR) of the band gap and temperature-dependent band energies within the harmonic approximation. We start with a very brief overview of the many-body formalism in the context of the electron-phonon (e-ph) interaction. Then we discuss how to evaluate the e-ph self-energy and perform typical convergence studies using the polar semiconductor MgO as an example. Further details concerning the implementation are given in [Gonze2019] and [Romero2020]. Additional examples are provided in this jupyter notebook that explains how to use Abipy to automate the calculations and post-process the results for Diamond.
It is assumed the user has already completed the two tutorials RF1 and RF2, and that he/she is familiar with the calculation of ground state (GS) and response properties in particular phonons, Born effective charges and the high-frequency dielectric tensor. The user should have read the introduction tutorial for the EPH code before running these examples.
This lesson should take about 1.5 hour.
Formalism¶
The electron-phonon self-energy, \Sigma^{\text{e-ph}}, describes the renormalization of charged electronic excitations due to the interaction with phonons. This term should be added to the electron-electron (e-e) self-energy \Sigma^{\text{e-e}} that encodes many-body effects induced by the Coulomb interaction beyond the classical electrostatic Hartree potential. The e-e contribution can be estimated using, for instance, the GW approximation but in this tutorial we are mainly interested in \Sigma^{\text{e-ph}} and its temperature dependence.
In semiconductors and insulators, indeed, most of the temperature dependence of the electronic properties at “low” T originates from the e-ph interaction and the thermal expansion of the unit cell (topic that, however, won’t be discussed in this lesson). Corrections due to \Sigma^{\text{e-e}} are obviously important as it is well known that KS gaps computed with LDA/GGA are systematically underestimated compared to experimental data. Nevertheless, the temperature dependence of \Sigma^{\text{e-e}} is rather small as long as kT is smaller than the fundamental gap (let’s say 3 kT < E_{\text{g}}).
In state-of-the-art ab-initio perturbative methods, the e-ph coupling is described within DFT by expanding the KS effective potential up to the second order in the atomic displacement, and the vibrational properties are computed with DFPT [Gonze1997], [Baroni2001]. Note that anharmonic effects that may become relevant at “high” T are not included in the present formalism.
The e-ph self-energy consists of two terms: the frequency-dependent Fan-Migdal (FM) self-energy and the static and Hermitian Debye-Waller (DW) part (see e.g. [Giustino2017] and references therein):
The diagonal matrix elements of the FM self-energy in the KS basis set are given by
where f_{m\kk+\qq}(\ef,T) and n_\qnu(T) are the Fermi-Dirac and Bose-Einstein occupation functions with T the physical temperature and \ef the Fermi level that in turns depends on T and the number of electrons per unit cell. The integration is performed over the \qq-points in the Brillouin zone (BZ) of volume \Omega_\BZ and \eta is a positive real infinitesimal.
Important
From a mathematical point of view, one should take the limit \eta \rightarrow 0^+. At the level of the implementation, the infinitesimal \eta is replaced by a (small) finite value given by the zcut variable that should be subject to convergence studies. More specifically, one should monitor the convergence of the physical properties of interest for zcut \rightarrow 0^+ and number of \qq-points \rightarrow \infty. Convergence studies should start from values of zcut that are comparable to the typical phonon frequency of the system (usually 0.01 eV or smaller). Note that the default value for zcut is 0.1 eV. This value is reasonable for GW calculations but 0.1 eV is, very likely, too large when computing \Sigma^{\text{e-ph}}.
The static DW term involves the second order derivative of the KS potential with respect to the nuclear displacements. State-of-the-art implementations approximate the DW contribution with
where g_{mn\nu}^{2,\DW}(\kk,\qq) is an effective matrix element that, within the rigid-ion approximation, can be expressed in terms of the standard first-order \gkq matrix elements by exploiting the invariance of the QP energies under infinitesimal translation [Giustino2017].
At the level of the implementation, the number of bands in the two sums is defined by nband while the \qq-mesh for the integration is specified by eph_ngqpt_fine (or ddb_ngqpt if the DFPT potentials are not interpolated). The list of temperatures (in Kelvin) is initialized from tmesh.
For the sake of simplicity, we will omit the T-dependence in the next equations. Keep in mind, however, that all the expressions in which \Sigma is involved have an additional dependence on the physical temperature T.
Important
The EPH code takes advantage of time-reversal and spatial symmetries to reduce the BZ integration to an appropriate irreducible wedge, \text{IBZ}_\kk, defined by the little group of the \kk-point i.e. the set of point group operations of the crystal that leave the \kk-point invariant within a reciprocal lattice vector \GG. Calculations at high-symmetry \kk-points such as \Gamma are therefore much faster as there are more symmetries that can be exploited (smaller \text{IBZ}_k).
This symmetrization procedure is activated by default and can be disabled by setting symsigma to 0 for testing purposes. Note that when symsigma is set to 1, the code performs a final average of the QP results within each degenerate subspace. As a consequence, accidental degeneracies won’t be removed when symsigma is set to 1.
Note that both the FM and the DW term converge slowly with the \qq-sampling. Moreover, accurate computation of the real part require the inclusion of a large number of empty states.
In order to accelerate the convergence with nband, the EPH code can replace the contributions given by the high-energy states above a certain band index M with the solution of a non-self-consistent Sternheimer equation in which only the first M states are required. The methodology, proposed in [Gonze2011], is based on a quasi-static approximation in which the phonon frequencies in the denominator of Eq. (\ref{eq:fan_selfen}) are neglected and the frequency dependence of \Sigma is approximated with the value computed at \omega = \enk. This approximation is justified when the bands above M are sufficiently high in energy with respect to the n\kk states that must be corrected. The Sternheimer approach requires the specification of eph_stern and getpot_filepath. The parameter M corresponds to the nband input variable that, obviously, cannot be larger than the number of bands stored in the WFK file (the code will abort if this condition is not fulfilled).
Quasi-particle corrections due to the e-ph coupling¶
Strictly speaking, the quasi-particle (QP) excitations are defined by the solution(s) in the complex plane of the equation $$ z = \ee_\nk + \Sigma_\nk^{\text{e-ph}}(z) $$ provided the non-diagonal components of the e-ph self-energy can be neglected. In practice, the problem is usually simplified by seeking approximated solutions along the real axis following two different approaches:
- on-the-mass-shell
- linearized QP equation.
In the on-the-mass-shell approximation, the QP energy is simply given by the real part of the self-energy evaluated at the bare KS eigenvalue:
In the linearized QP equation, on the contrary, the self-energy is Taylor-expanded around the bare KS eigenvalue and the QP correction is obtained using
with the renormalization factor Z_\nk given by
Note that Z_\nk is approximately equal to the area under the QP peak in the spectral function:
Since A_\nk(\ww) integrates to 1, values of Z_\nk in the [0.7, 1] range usually indicate the presence of a well-defined QP excitation that may be accompanied by some background and, possibly, additional satellites. Values of Z_\nk greater than one are clearly unphysical and signal the breakdown of the linearized QP equation. The interpretation of these results requires a careful analysis of A_\nk(\ww) and/or additional convergence tests.
Important
Both approaches are implemented in ABINIT although it should be noted that, according to recent works, the on-the-mass-shell approach provides results that are closer to those obtained with more advanced techniques based on the cumulant expansion [Nery2018].
The ZPR of the excitation energy is defined as the difference between the QP energy evaluated at T = 0 and the bare KS energy. In a similar way, one defines the ZPR of the gap as the difference between the QP band gap at T = 0 and the KS gap.
It is worth to stress that the EPH code can compute QP corrections only for \nk states that are present in the input WFK file (a similar requirement is present in the GW code as well). As a consequence, the \kk-mesh (ngkpt, nshiftk, shiftk) for the WFK file should be chosen carefully especially if the band edges are not located at high-symmetry \kk-points.
Note that there are different approaches one can use to specify the set of \nk states in \Sigma_{\nk}. Each approach has pros and cons. The most direct way consists in specifying the \kk-points and the band range using the three variables: nkptgw, kptgw, bdgw For instance, in order to compute the correction for the VBM/CBM at \Gamma in silicon (non-magnetic semiconductor with 8 valence electrons per unit cell), one would use:
nkptgw 1 kptgw 0 0 0 # [3, nkptgw] array bdgw 4 5 # [2, nkptgw] array giving the initial and the last band index # for each nkptgw k-point
as the index of the valence band is given by 8 / 2 = 4. Obviously, this input file can only provide the ZPR of the direct gap as Si has an indirect fundamental gap. This the most flexible approach but it requires the specification of three variables and, obviously, one should know the positions of the CBM/VBM. Alternatively, one can use gw_qprange or sigma_erange.
Important
When symsigma is set to 1 (default), the code may decide to enlarge the initial value of bdgw so that all degenerate states for that particular \kk-point are included in the calculation.
Typical workflow for ZPR¶
A typical workflow for ZPR calculations involves the following steps (see the introductory e-ph tutorial):
-
GS calculation to obtain the WFK and the DEN file. The \kk-mesh should be dense enough to converge both electronic and vibrational properties. Remember to set prtpot to 1 to produce the file with the KS potential required by the Sternheimer method.
-
DFPT calculations for all the IBZ \qq-points corresponding to the ab-initio ddb_ngqpt mesh that will be used to perform the Fourier interpolation of the dynamical matrix and of the DFPT potentials. In the simplest case, one uses a \qq-mesh that is equal to the GS \kk-mesh (sub-meshes are also fine) and the DFPT calculations can directly start from the WFK produced in step #1. Remember to compute \bm{\epsilon}^{\infty}, \bm{Z}^* (polar materials) and the dynamical quadrupoles \bm{Q}^* as these quantities are needed for an accurate interpolation of phonon frequencies and DFPT potentials.
-
NSCF computation of a WFK file on a much denser \kk-mesh containing the wavevectors where phonon-induced QP corrections are wanted. The NSCF run uses the DEN file produced in step #1. Remember to compute enough empty states so that it is possible to perform convergence studies w.r.t. nband afterwards.
-
Merge the partial DDB and POT files with mrgddb and mrgdvdb, respectively.
-
Start from the DDB/DVDB files produced in step #4 and the WFK file obtained in step #3 to perform ZPR calculations with eph_task 4.
Getting started¶
Note
Supposing you made your own installation of ABINIT, the input files to run the examples are in the ~abinit/tests/ directory where ~abinit is the absolute path of the abinit top-level directory. If you have NOT made your own install, ask your system administrator where to find the package, especially the executable and test files.
In case you work on your own PC or workstation, to make things easier, we suggest you define some handy environment variables by executing the following lines in the terminal:
export ABI_HOME=Replace_with_absolute_path_to_abinit_top_level_dir # Change this line export PATH=$ABI_HOME/src/98_main/:$PATH # Do not change this line: path to executable export ABI_TESTS=$ABI_HOME/tests/ # Do not change this line: path to tests dir export ABI_PSPDIR=$ABI_TESTS/Psps_for_tests/ # Do not change this line: path to pseudos dir
Examples in this tutorial use these shell variables: copy and paste
the code snippets into the terminal (remember to set ABI_HOME first!) or, alternatively,
source the set_abienv.sh script located in the ~abinit directory:
source ~abinit/set_abienv.sh
The ‘export PATH’ line adds the directory containing the executables to your PATH so that you can invoke the code by simply typing abinit in the terminal instead of providing the absolute path.
To execute the tutorials, create a working directory (Work*) and
copy there the input files of the lesson.
Most of the tutorials do not rely on parallelism (except specific tutorials on parallelism). However you can run most of the tutorial examples in parallel with MPI, see the topic on parallelism.
Before beginning, you might consider to work in a different subdirectory as for the other tutorials. Why not create Work_eph4zpr in $ABI_TESTS/tutorespfn/Input?
cd $ABI_TESTS/tutorespfn/Input mkdir Work_eph4zpr cd Work_eph4zpr
In this tutorial, we prefer to focus on the use of the EPH code hence we will be using pre-computed DDB and DFPT POT files to bypass the DFPT part. We also provide a DEN.nc file to initialize the NSCF calculations and a POT file with the GS KS potential required to solve the Sternheimer equation.
If git is installed on your machine, one can easily fetch the entire repository (23 MB) with:
git clone https://github.com/abinit/MgO_eph_zpr.git
Alternatively, use wget:
wget https://github.com/abinit/MgO_eph_zpr/archive/master.zip
or curl:
curl -L https://github.com/abinit/MgO_eph_zpr/archive/master.zip -o master.zip
or simply copy the tarball by clicking the “download button” available in the github web page, unzip the file and rename the directory with:
unzip master.zip mv MgO_eph_zpr-master MgO_eph_zpr
Warning
The directory with the precomputed files must be located in the same working directory
in which you will be executing the tutorial and must be named MgO_eph_zpr.
The AbiPy script used to executed the DFPT part is available here. Note that several parameters have been tuned to reach a reasonable compromise between accuracy and computational cost so do not expect the results obtained at the end of the lesson to be fully converged. More specifically, we use norm-conserving pseudopotentials with a cutoff energy ecut of 30 Ha (too low, it should be ~50 Ha). The DFPT computations is done for the set of irreducible \qq-points corresponding to a \Gamma-centered 4x4x4 \qq mesh (again, too coarse). \bm{Z}^* and \bm{\ee}^\infty are also computed with the same underconverged settings.
Since AbiPy does not support multiple datasets, each directory corresponds to a single calculation.
In particular, all the DFPT tasks (atomic perturbations, DDK, electric field perturbation)
can be found inside the w1 directory while w0/t0/outdata contains the GS results.
and w0/t1/outdata the GSR file with energie on an high-symmetry \kk-path.
How to extract useful info from the output files¶
Since this is the first tutorial that uses precomputed output files, it is worth explaining how to use the terminal and command line utilities such as ncdump, abitk and AbiPy scripts to extract useful information from the pre-computed files before moving to EPH calculations.
First of all, most of the netcdf files produced by ABINIT store the input file in string format in the input_string netcdf variable. This variable may be useful if you need to know the input variables used to produce that particular output file. To print the value of input_string inside the terminal, use the ncdump utility and the syntax:
ncdump -v input_string MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc input_string = "jdtset 1 nband 12 ecut 35.0 ngkpt 4 4 4 nshiftk 1 shiftk 0 0 0 tolvrs 1e-12 nstep 150 iomode 3 prtpot 1 diemac 9.0 nbdbuf 4 paral_kgb 0 natom 2 ntypat 2 typat 2 1 znucl 8 12 xred 0.0000000000 0.0000000000 0.0000000000 0.5000000000 0.5000000000 0.5000000000 acell 1.0 1.0 1.0 rprim 0.0000000000 4.0182361526 4.0182361526 4.0182361526 0.0000000000 4.0182361526 4.0182361526 4.0182361526 0.0000000000" ;
In all the examples of this tutorial, we will be using the new structure input variable (added in version 9) to initialize the unit cell from an external file so that we don’t need to repeat the unit cell over and over again in all the input files. The syntax is :
structure "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc"
where the abifile prefix tells ABINIT that the lattice parameters and atomic positions
should be extracted from an ABINIT binary file e.g. HIST.nc, DEN.nc, GSR.nc, etc.
(other formats are supported as well, see the documentation).
To print the crystalline structure to terminal, use the abitk Fortran executable
shipped with the ABINIT package and the crystal_print command:
abitk crystal_print MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc
==== Info on the Cryst% object ====
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Time-reversal symmetry is present
Reduced atomic positions [iatom, xred, symbol]:
1) 0.0000000 0.0000000 0.0000000 Mg
2) 0.5000000 0.5000000 0.5000000 O
This is the crystalline structure that we will be using in the forthcoming examples.
Since we want to compute the renormalization of the band gap due to phonons, it is also useful to have a look at the KS gaps obtained from the GS run. The command is:
abitk ebands_gaps MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc Direct band gap semiconductor Fundamental gap: 4.479 (eV) VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00] CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00] Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
The same abitk command can be used with all netcdf files containing KS energies e.g GSR.nc, WFK.nc.
Warning
Our values for the gaps are consistent with the results for MgO given on the materialsproject. Remember, however, that the values and the positions of the gaps may vary (in somes cases even significantly) depending on the \kk-sampling.
In this case, abitk reports the gaps computed from a \kk-mesh as the DEN file can only be produced by a SCF calculation. The results for MgO are correct simply because the CBM/VBM are at the \Gamma point and this point belongs to the GS \kk-mesh. Other systems (e.g. Si) may have the CBM/VBM at wavevectors that are not easily captured with a homogeneous mesh. The most reliable approach to find the location of the CBM/VBM is to perform a band structure calculation on a high-symmetry \kk-path.
abitk is handy if you need to call Fortran routines from the terminal to perform basic tasks
but Fortran is not the best language when it comes to post-processing and data analysis.
This kind of operation, indeed, is much easier to implement using a high-level language such as python.
To plot the band structure using the GS eigenvalues stored in the GSR.nc file,
use the abiopen script provided by AbiPy with the -e option:
abiopen.py MgO_eph_zpr/flow_zpr_mgo/w0/t1/outdata/out_GSR.nc -e
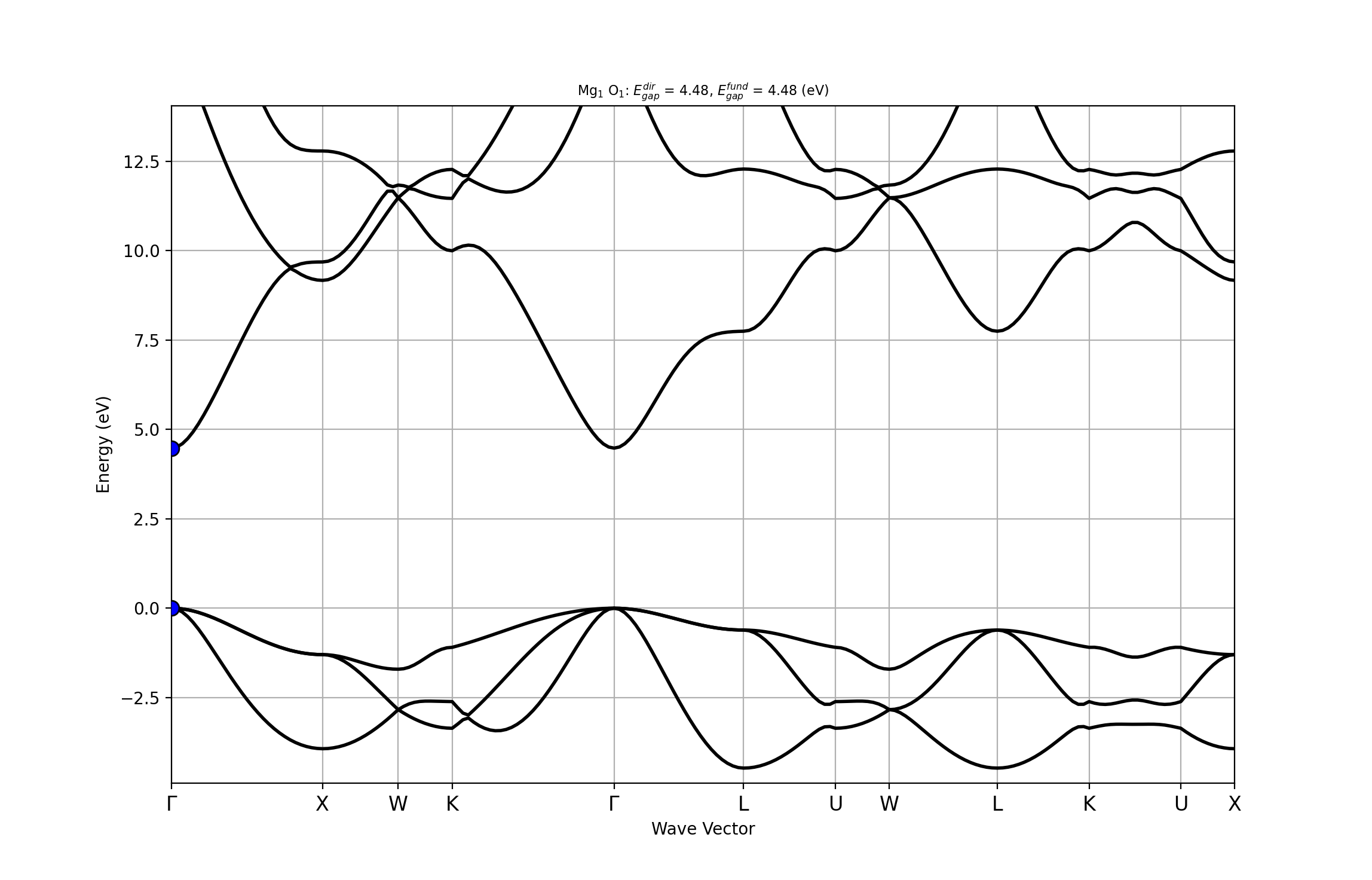
The figure confirms that the gap is direct at the \Gamma point. The VBM is three-fold degenerate when SOC is not included.
How to merge partial DDB files with mrgddb¶
First of all, let’s merge the partial DDB files with the command
mrgddb < teph4zpr_1.abi
and the following input file:
teph4zpr_1_DDB MgO phonons on 4x4x4 q-mesh 28 MgO_eph_zpr/flow_zpr_mgo/w1/t3/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t4/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t5/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t6/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t7/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t8/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t9/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t10/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t11/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t12/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t13/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t14/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t15/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t16/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t17/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t18/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t19/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t20/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t21/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t22/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t23/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t24/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t25/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t26/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t27/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t28/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t29/outdata/out_DDB MgO_eph_zpr/flow_zpr_mgo/w1/t30/outdata/out_DDB ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = mrgddb #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_1.stdout, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 1 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = Merge precomputed DDB files stored in the MgO_eph_zpr git submodule #%%<END TEST_INFO>
that lists the relative paths of the partial DDB files in the
MgO_eph_zpr directory.
Since we are dealing with a polar material, it is worth checking whether our final DDB contains
Born effective charges and the electronic dielectric tensor.
Instead of running anaddb or abinit and then checking the output file,
we can simply use abiopen with the -p option:
abiopen.py MgO_eph_zpr/flow_zpr_mgo/w1//outdata/out_DDB -p ================================== DDB Info ================================== Number of q-points in DDB: 8 guessed_ngqpt: [4 4 4] (guess for the q-mesh divisions made by AbiPy) ecut = 35.000000, ecutsm = 0.000000, nkpt = 36, nsym = 48, usepaw = 0 nsppol 1, nspinor 1, nspden 1, ixc = 11, occopt = 1, tsmear = 0.010000 Has total energy: False, Has forces: False Has stress tensor: False Has (at least one) atomic pertubation: True Has (at least one diagonal) electric-field perturbation: True Has (at least one) Born effective charge: True Has (all) strain terms: False Has (all) internal strain terms: False Has (all) piezoelectric terms: False
We can also invoke anaddb directly from python to have a quick look at the phonon dispersion:
abiview.py ddb MgO_eph_zpr/flow_zpr_mgo/w1//outdata/out_DDB Computing phonon bands and DOS from DDB file with nqsmall = 10, ndivsm = 20; asr = 2, chneut = 1, dipdip = 1, lo_to_splitting = automatic, dos_method = tetra
that produces the following figures:
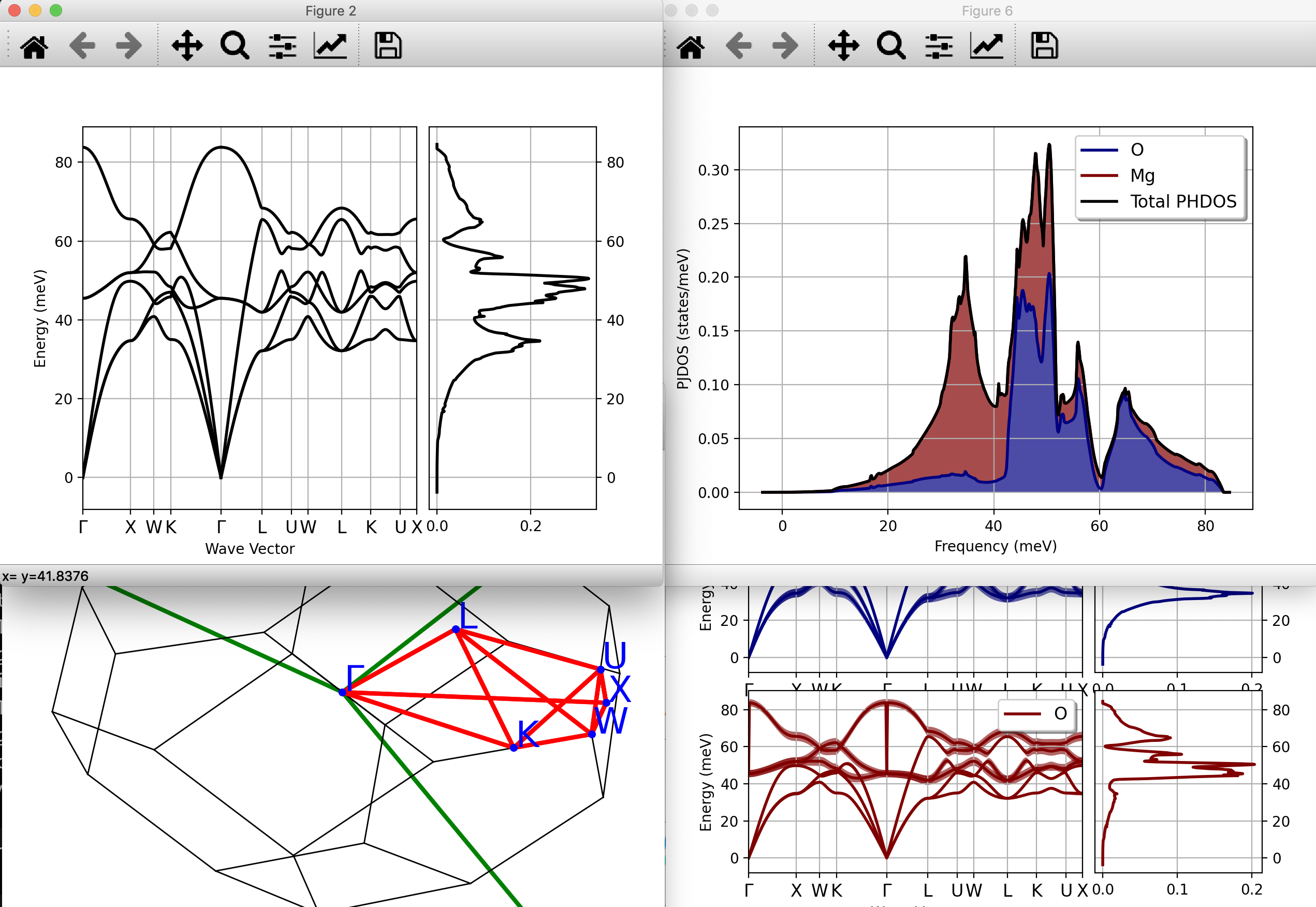
The results seem reasonable: the acoustic modes go to zero linearly for \qq \rightarrow 0 as we are dealing with a 3D system, no instability is present and the phonon dispersion shows the LO-TO splitting typical of polar materials.
Note, however, that the acoustic sum-rule is automatically enforced by the code so it is always a good idea to compare the results with/without asr as this is an indirect indicator of the convergence/reliability of our calculations. We can automate the process with the ddb_asr command of abiview :
abiview.py ddb_asr MgO_eph_zpr/flow_zpr_mgo/w1//outdata/out_DDB
that produces the following figure:
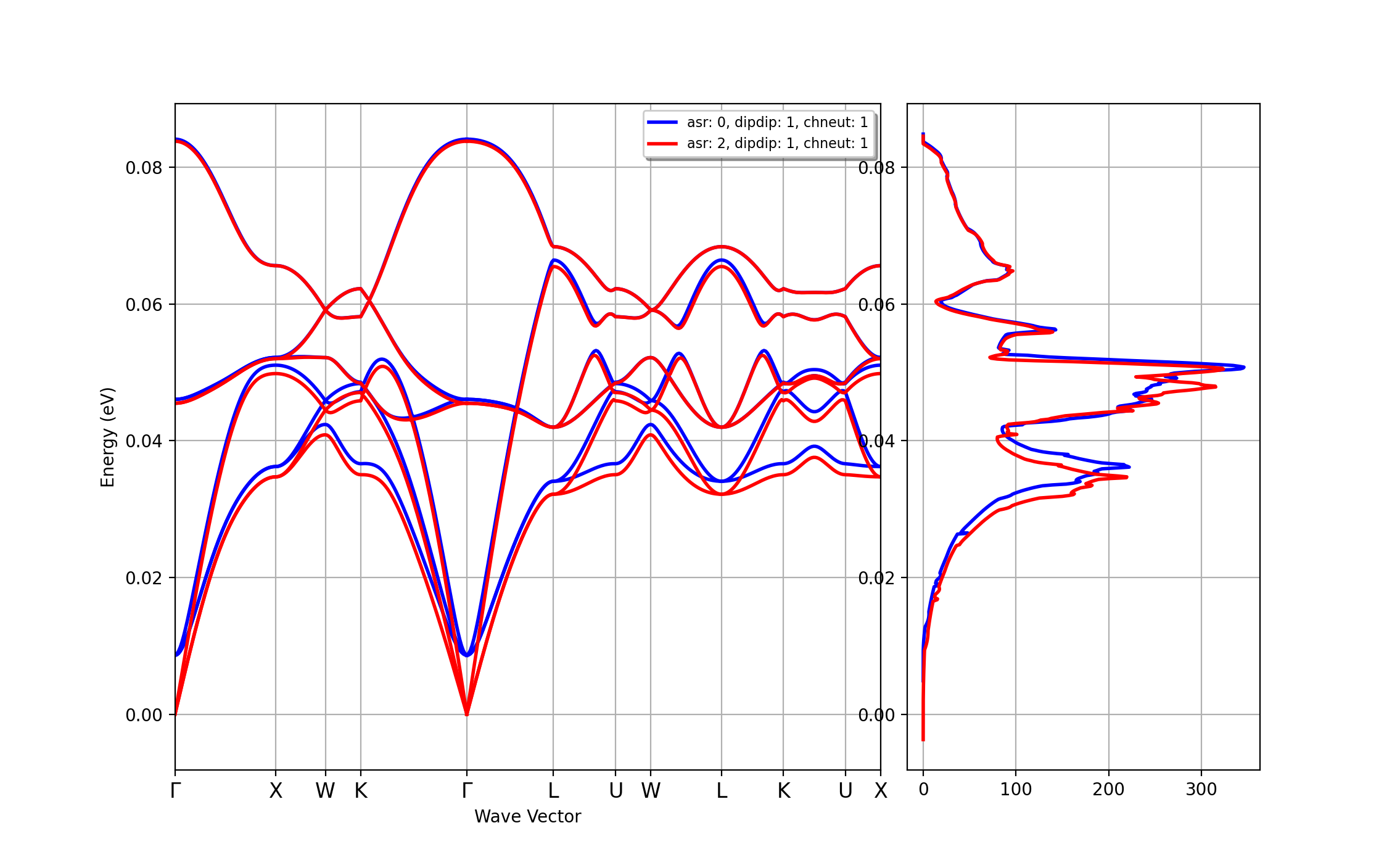
Important
This clearly indicates that the breaking of the acoustic sum-rule is not negligible. In this case, the breaking is mainly due the too low cutoff energy employed in our calculations. In real life, one should stop here and redo the DFPT calculation with a larger ecut and possibly a denser \qq-mesh but since the goal of this lesson is to teach you how to run ZPR calculations, we ignore this serious problem and continue with the other examples.
PS: If you want to compute the phonons bands with/without dipdip, use:
abiview.py ddb_dipdip MgO_eph_zpr/flow_zpr_mgo/w1//outdata/out_DDB
How to merge partial POT files with mrgdv¶
Now we can merge the DFPT potential with the mrgdv tool using the command.
mrgdv < teph4zpr_2.abi
and the following input file:
teph4zpr_2_DVDB 28 MgO_eph_zpr/flow_zpr_mgo/w1/t3/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t4/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t5/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t6/outdata/out_POT2.nc MgO_eph_zpr/flow_zpr_mgo/w1/t7/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t8/outdata/out_POT5.nc MgO_eph_zpr/flow_zpr_mgo/w1/t9/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t10/outdata/out_POT2.nc MgO_eph_zpr/flow_zpr_mgo/w1/t11/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t12/outdata/out_POT5.nc MgO_eph_zpr/flow_zpr_mgo/w1/t13/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t14/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t15/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t16/outdata/out_POT2.nc MgO_eph_zpr/flow_zpr_mgo/w1/t17/outdata/out_POT3.nc MgO_eph_zpr/flow_zpr_mgo/w1/t18/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t19/outdata/out_POT5.nc MgO_eph_zpr/flow_zpr_mgo/w1/t20/outdata/out_POT6.nc MgO_eph_zpr/flow_zpr_mgo/w1/t21/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t22/outdata/out_POT3.nc MgO_eph_zpr/flow_zpr_mgo/w1/t23/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t24/outdata/out_POT6.nc MgO_eph_zpr/flow_zpr_mgo/w1/t25/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t26/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t27/outdata/out_POT1.nc MgO_eph_zpr/flow_zpr_mgo/w1/t28/outdata/out_POT2.nc MgO_eph_zpr/flow_zpr_mgo/w1/t29/outdata/out_POT4.nc MgO_eph_zpr/flow_zpr_mgo/w1/t30/outdata/out_POT5.nc ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = mrgdv #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_2.stdout, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 1 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = Merge precomputed DFPT POT files stored in the MgO_eph_zpr git submodule #%%<END TEST_INFO>
Tip
The number at the end of the POT file corresponds to the (idir, ipert) pertubation for that particular \qq-point. The pertcase index is computed as:
pertcase = idir + ipert
where idir gives the direction ([1, 2, 3]) and ipert specifies the perturbation type:
- ipert in [1, …, natom] corresponds to atomic perturbations (reduced directions)
- ipert = natom + 1 corresponds d/dk (reduced directions)
- ipert = natom + 2 corresponds the electric field
- ipert = natom + 3 corresponds the uniaxial stress (Cartesian directions)
- ipert = natom + 4 corresponds the shear stress. (Cartesian directions)
All DFPT POT files with 1 <= pertcase <= 3 x natom therefore correspond to atomic pertubations for a given \qq-point.
The value of pertcase and qpt are reported in the ABINIT header. To print the header to terminal, use abitk with the hdr_print command
abitk hdr_print MgO_eph_zpr/flow_zpr_mgo/w1/t11/outdata/out_POT4.nc =============================================================================== ECHO of part of the ABINIT file header First record : .codvsn,headform,fform = 9.2.0 80 111 Second record : bantot,intxc,ixc,natom = 480 0 11 2 ngfft(1:3),nkpt = 32 32 32 40 nspden,nspinor = 1 1 nsppol,nsym,npsp,ntypat = 1 48 2 2 occopt,pertcase,usepaw = 1 4 0 ecut,ecutdg,ecutsm = 3.5000000000E+01 3.5000000000E+01 0.0000000000E+00 ecut_eff = 3.5000000000E+01 qptn(1:3) = 5.0000000000E-01 0.0000000000E+00 0.0000000000E+00 rprimd(1:3,1) = 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00 rprimd(1:3,2) = 4.0182361526E+00 0.0000000000E+00 4.0182361526E+00 rprimd(1:3,3) = 4.0182361526E+00 4.0182361526E+00 0.0000000000E+00 stmbias,tphysel,tsmear = 0.0000000000E+00 0.0000000000E+00 1.0000000000E-02 The header contain 4 additional records.
Use --prtvol 1 to output more records.
Now we discuss in more detail the output file produced by mrgdv
.Version 9.3.1 of MRGDV
.(MPI version, prepared for a x86_64_linux_gnu9.3 computer)
.Copyright (C) 1998-2021 ABINIT group .
MRGDV comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Tue 10 Nov 2020.
- ( at 22h19 )
->> Enter name of output file:
->> Enter total number of DFPT POT files:
->> Enter name of POT file 1 :
->> Enter name of POT file 2 :
->> Enter name of POT file 3 :
->> Enter name of POT file 4 :
->> Enter name of POT file 5 :
->> Enter name of POT file 6 :
->> Enter name of POT file 7 :
->> Enter name of POT file 8 :
->> Enter name of POT file 9 :
->> Enter name of POT file 10 :
->> Enter name of POT file 11 :
->> Enter name of POT file 12 :
->> Enter name of POT file 13 :
->> Enter name of POT file 14 :
->> Enter name of POT file 15 :
->> Enter name of POT file 16 :
->> Enter name of POT file 17 :
->> Enter name of POT file 18 :
->> Enter name of POT file 19 :
->> Enter name of POT file 20 :
->> Enter name of POT file 21 :
->> Enter name of POT file 22 :
->> Enter name of POT file 23 :
->> Enter name of POT file 24 :
->> Enter name of POT file 25 :
->> Enter name of POT file 26 :
->> Enter name of POT file 27 :
->> Enter name of POT file 28 :
- Reading header of file [1]: MgO_eph_zpr/flow_zpr_mgo/w1/t3/outdata/out_POT1.nc
- Merging file [1]: MgO_eph_zpr/flow_zpr_mgo/w1/t3/outdata/out_POT1.nc
- Reading header of file [2]: MgO_eph_zpr/flow_zpr_mgo/w1/t4/outdata/out_POT4.nc
- Merging file [2]: MgO_eph_zpr/flow_zpr_mgo/w1/t4/outdata/out_POT4.nc
- Reading header of file [3]: MgO_eph_zpr/flow_zpr_mgo/w1/t5/outdata/out_POT1.nc
- Merging file [3]: MgO_eph_zpr/flow_zpr_mgo/w1/t5/outdata/out_POT1.nc
- Reading header of file [4]: MgO_eph_zpr/flow_zpr_mgo/w1/t6/outdata/out_POT2.nc
- Merging file [4]: MgO_eph_zpr/flow_zpr_mgo/w1/t6/outdata/out_POT2.nc
- Reading header of file [5]: MgO_eph_zpr/flow_zpr_mgo/w1/t7/outdata/out_POT4.nc
- Merging file [5]: MgO_eph_zpr/flow_zpr_mgo/w1/t7/outdata/out_POT4.nc
- Reading header of file [6]: MgO_eph_zpr/flow_zpr_mgo/w1/t8/outdata/out_POT5.nc
- Merging file [6]: MgO_eph_zpr/flow_zpr_mgo/w1/t8/outdata/out_POT5.nc
- Reading header of file [7]: MgO_eph_zpr/flow_zpr_mgo/w1/t9/outdata/out_POT1.nc
- Merging file [7]: MgO_eph_zpr/flow_zpr_mgo/w1/t9/outdata/out_POT1.nc
- Reading header of file [8]: MgO_eph_zpr/flow_zpr_mgo/w1/t10/outdata/out_POT2.nc
- Merging file [8]: MgO_eph_zpr/flow_zpr_mgo/w1/t10/outdata/out_POT2.nc
- Reading header of file [9]: MgO_eph_zpr/flow_zpr_mgo/w1/t11/outdata/out_POT4.nc
- Merging file [9]: MgO_eph_zpr/flow_zpr_mgo/w1/t11/outdata/out_POT4.nc
- Reading header of file [10]: MgO_eph_zpr/flow_zpr_mgo/w1/t12/outdata/out_POT5.nc
- Merging file [10]: MgO_eph_zpr/flow_zpr_mgo/w1/t12/outdata/out_POT5.nc
- Reading header of file [11]: MgO_eph_zpr/flow_zpr_mgo/w1/t13/outdata/out_POT1.nc
- Merging file [11]: MgO_eph_zpr/flow_zpr_mgo/w1/t13/outdata/out_POT1.nc
- Reading header of file [12]: MgO_eph_zpr/flow_zpr_mgo/w1/t14/outdata/out_POT4.nc
- Merging file [12]: MgO_eph_zpr/flow_zpr_mgo/w1/t14/outdata/out_POT4.nc
- Reading header of file [13]: MgO_eph_zpr/flow_zpr_mgo/w1/t15/outdata/out_POT1.nc
- Merging file [13]: MgO_eph_zpr/flow_zpr_mgo/w1/t15/outdata/out_POT1.nc
- Reading header of file [14]: MgO_eph_zpr/flow_zpr_mgo/w1/t16/outdata/out_POT2.nc
- Merging file [14]: MgO_eph_zpr/flow_zpr_mgo/w1/t16/outdata/out_POT2.nc
- Reading header of file [15]: MgO_eph_zpr/flow_zpr_mgo/w1/t17/outdata/out_POT3.nc
- Merging file [15]: MgO_eph_zpr/flow_zpr_mgo/w1/t17/outdata/out_POT3.nc
- Reading header of file [16]: MgO_eph_zpr/flow_zpr_mgo/w1/t18/outdata/out_POT4.nc
- Merging file [16]: MgO_eph_zpr/flow_zpr_mgo/w1/t18/outdata/out_POT4.nc
- Reading header of file [17]: MgO_eph_zpr/flow_zpr_mgo/w1/t19/outdata/out_POT5.nc
- Merging file [17]: MgO_eph_zpr/flow_zpr_mgo/w1/t19/outdata/out_POT5.nc
- Reading header of file [18]: MgO_eph_zpr/flow_zpr_mgo/w1/t20/outdata/out_POT6.nc
- Merging file [18]: MgO_eph_zpr/flow_zpr_mgo/w1/t20/outdata/out_POT6.nc
- Reading header of file [19]: MgO_eph_zpr/flow_zpr_mgo/w1/t21/outdata/out_POT1.nc
- Merging file [19]: MgO_eph_zpr/flow_zpr_mgo/w1/t21/outdata/out_POT1.nc
- Reading header of file [20]: MgO_eph_zpr/flow_zpr_mgo/w1/t22/outdata/out_POT3.nc
- Merging file [20]: MgO_eph_zpr/flow_zpr_mgo/w1/t22/outdata/out_POT3.nc
- Reading header of file [21]: MgO_eph_zpr/flow_zpr_mgo/w1/t23/outdata/out_POT4.nc
- Merging file [21]: MgO_eph_zpr/flow_zpr_mgo/w1/t23/outdata/out_POT4.nc
- Reading header of file [22]: MgO_eph_zpr/flow_zpr_mgo/w1/t24/outdata/out_POT6.nc
- Merging file [22]: MgO_eph_zpr/flow_zpr_mgo/w1/t24/outdata/out_POT6.nc
- Reading header of file [23]: MgO_eph_zpr/flow_zpr_mgo/w1/t25/outdata/out_POT1.nc
- Merging file [23]: MgO_eph_zpr/flow_zpr_mgo/w1/t25/outdata/out_POT1.nc
- Reading header of file [24]: MgO_eph_zpr/flow_zpr_mgo/w1/t26/outdata/out_POT4.nc
- Merging file [24]: MgO_eph_zpr/flow_zpr_mgo/w1/t26/outdata/out_POT4.nc
- Reading header of file [25]: MgO_eph_zpr/flow_zpr_mgo/w1/t27/outdata/out_POT1.nc
- Merging file [25]: MgO_eph_zpr/flow_zpr_mgo/w1/t27/outdata/out_POT1.nc
- Reading header of file [26]: MgO_eph_zpr/flow_zpr_mgo/w1/t28/outdata/out_POT2.nc
- Merging file [26]: MgO_eph_zpr/flow_zpr_mgo/w1/t28/outdata/out_POT2.nc
- Reading header of file [27]: MgO_eph_zpr/flow_zpr_mgo/w1/t29/outdata/out_POT4.nc
- Merging file [27]: MgO_eph_zpr/flow_zpr_mgo/w1/t29/outdata/out_POT4.nc
- Reading header of file [28]: MgO_eph_zpr/flow_zpr_mgo/w1/t30/outdata/out_POT5.nc
- Merging file [28]: MgO_eph_zpr/flow_zpr_mgo/w1/t30/outdata/out_POT5.nc
Merged successfully 28 files
- Analyzing DVDB file: teph4zpr_2_DVDB ...
- dvdb_new completed. cpu: 0.00 [s] , wall: 0.00 [s] <<< TIME
==== Info on the dvdb% object ====
DVDB version: 2
File path: teph4zpr_2_DVDB
Number of v1scf potentials: 28
Number of q-points in DVDB: 8
-P Number of CPUs for parallelism over perturbations: 1
-P Number of perturbations treated by this CPU: 6
Option for symmetrization of v1scf(r): 0
List of q-points: min(10, nqpt)
[ 1 ] [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
[ 2 ] [ 2.5000E-01, 0.0000E+00, 0.0000E+00]
[ 3 ] [ 5.0000E-01, 0.0000E+00, 0.0000E+00]
[ 4 ] [ 2.5000E-01, 2.5000E-01, 0.0000E+00]
[ 5 ] [ 5.0000E-01, 2.5000E-01, 0.0000E+00]
[ 6 ] [-2.5000E-01, 2.5000E-01, 0.0000E+00]
[ 7 ] [ 5.0000E-01, 5.0000E-01, 0.0000E+00]
[ 8 ] [-2.5000E-01, 5.0000E-01, 2.5000E-01]
Have dielectric tensor: no
Have Born effective charges: no
Have quadrupoles: no
Have electric field: no
Treatment of long-range part in V1scf (add_lr): 1
Damping factor for Gaussian filter (qdamp): 0.1
qpoint: [ 0.0000E+00, 0.0000E+00, 0.0000E+00] is present in the DVDB file
The list of irreducible perturbations for this q vector is:
1) idir= 1, ipert= 1, type=independent, found=Yes
2) idir= 2, ipert= 1, type=symmetric, found=No
3) idir= 3, ipert= 1, type=symmetric, found=No
4) idir= 1, ipert= 2, type=independent, found=Yes
5) idir= 2, ipert= 2, type=symmetric, found=No
6) idir= 3, ipert= 2, type=symmetric, found=No
qpoint: [ 2.5000E-01, 0.0000E+00, 0.0000E+00] is present in the DVDB file
The list of irreducible perturbations for this q vector is:
1) idir= 1, ipert= 1, type=independent, found=Yes
2) idir= 2, ipert= 1, type=independent, found=Yes
3) idir= 3, ipert= 1, type=symmetric, found=No
4) idir= 1, ipert= 2, type=independent, found=Yes
5) idir= 2, ipert= 2, type=independent, found=Yes
6) idir= 3, ipert= 2, type=symmetric, found=No
qpoint: [ 5.0000E-01, 0.0000E+00, 0.0000E+00] is present in the DVDB file
The list of irreducible perturbations for this q vector is:
1) idir= 1, ipert= 1, type=independent, found=Yes
2) idir= 2, ipert= 1, type=independent, found=Yes
3) idir= 3, ipert= 1, type=symmetric, found=No
4) idir= 1, ipert= 2, type=independent, found=Yes
5) idir= 2, ipert= 2, type=independent, found=Yes
6) idir= 3, ipert= 2, type=symmetric, found=No
qpoint: [ 2.5000E-01, 2.5000E-01, 0.0000E+00] is present in the DVDB file
The list of irreducible perturbations for this q vector is:
1) idir= 1, ipert= 1, type=independent, found=Yes
2) idir= 2, ipert= 1, type=symmetric, found=No
3) idir= 3, ipert= 1, type=symmetric, found=No
4) idir= 1, ipert= 2, type=independent, found=Yes
5) idir= 2, ipert= 2, type=symmetric, found=No
6) idir= 3, ipert= 2, type=symmetric, found=No
More than 20 q-points with prtvol == 0. Only important messages will be printed...
qpoint: [ 5.0000E-01, 2.5000E-01, 0.0000E+00] is present in the DVDB file
The list of irreducible perturbations for this q vector is:
1) idir= 1, ipert= 1, type=independent, found=Yes
2) idir= 2, ipert= 1, type=independent, found=Yes
3) idir= 3, ipert= 1, type=independent, found=Yes
4) idir= 1, ipert= 2, type=independent, found=Yes
5) idir= 2, ipert= 2, type=independent, found=Yes
6) idir= 3, ipert= 2, type=independent, found=Yes
All the independent perturbations are available
Done
For each \qq-point found in the partial POT files, the code prints a lists with the atomic perturbations that have been merged in the database.
qpoint: [ 0.0000E+00, 0.0000E+00, 0.0000E+00] is present in the DVDB file
The list of irreducible perturbations for this q vector is:
1) idir= 1, ipert= 1, type=independent, found=Yes
2) idir= 2, ipert= 1, type=symmetric, found=No
3) idir= 3, ipert= 1, type=symmetric, found=No
4) idir= 1, ipert= 2, type=independent, found=Yes
5) idir= 2, ipert= 2, type=symmetric, found=No
6) idir= 3, ipert= 2, type=symmetric, found=No
The term symmetric means that this particular (idir, ipert) perturbation can be reconstructed by symmetry from the other independent entries with the same \qq-point. If all the independent entries are available, the code prints the following message at the end of the output file:
All the independent perturbations are available Done
Warning
If you don’t get this message, the DVDB cannot be used by the EPH code. In this case, check carefully your DFPT input files and the list of POT files that have been merged. Also, note that it is not possible to change the value of nsym at the level of the EPH calculation as symmetries are automatically inherited from the previous GS/DFPT calculations.
Computing the WFK files with empty states¶
At this point we have all the ingredients (DDB and DVDB) required to compute/interpolate the e-ph scattering potentials and we can finally start to generate the WFK files.
For our first NSCF calculation, we use a 4x4x4 \Gamma-centered \kk-mesh and 70 bands so that we can perform initial convergence studies for the number of empty states in the self-energy. Then we generate WFK files with denser meshes and less bands that will be used for the Sternheimer method. Note the use of getden_filepath to read the DEN.nc file instead of getden or irdden.
You may now run the NSCF calculation by issuing:
abinit teph4zpr_3.abi > teph4zpr_3.log 2> err &
with the input file given by:
# NSCF computation of WFK files with empty states to prepare convergence studies for ZPR calculation ndtset 3 nband1 70 nbdbuf1 -10 # A negative value means 10% of nband nband 25 nbdbuf 5 # Three Gamma-centered k-meshes: # 4x4x4 with 70 bands, then 8x8x8 and 12x12x12 with 25 bands. ngkpt1 4 4 4 ngkpt2 8 8 8 getwfk2 -1 # Read previous WFK file to speedup NSCF run. ngkpt3 12 12 12 getwfk3 -1 nshiftk 1 shiftk 0 0 0 ######################## # Common to all datasets ######################## ecut 35.0 nstep 150 iscf -2 # NSCF run tolwfr 1e-14 # 1e-18 is the recommended value getden_filepath "MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" structure "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" pp_dirpath "$ABI_PSPDIR" pseudos "O.psp8, Mg-sp-gw.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_3.abo, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 10 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = NSCF computation of WFK files with empty states to prepare convergence studies for ZPR calculation #%%<END TEST_INFO>
At this point, it is worth commenting about the use of nbdbuf. As mentioned in the documentation, the highest energy states require more iterations to convergence. To avoid wasting precious computing time, we use a buffer that is ~10% of nband. This tricks significantly reduces the wall-time as the NSCF calculation completes when only the first nband - nbdbuf states are converged within tolwfr. Obviously, one should not use the last nbdbuf states in the subsequent EPH calculation. The same trick is highly recommended when computing WFK files for GW calculations.
Important
For mobility calculations, it is possible to reduce significantly the cost of the WFK computation by restricting the NSCF calculation to the \kk-points inside the electron (hole) pockets relevant for transport. Unfortunately, this optimization is not possible when computing the real part of the self-energy as the integration must be performed in the full \text{IBZ}_\kk. On the other hand, ZPR calculations can take advange of the Sternheimer method to reduce the number of empty bands required to converge.
Our first ZPR calculation¶
For our first example, we use a minimalistic input file so that we can discuss the most important input variables and the content of the main output file. First of all, you may want to start immediately the computation by issuing:
abinit teph4zpr_4.abi > teph4zpr_4.log 2> err &
with the following input file:
# ZPR computation at the Gamma point for MgO. nband 30 ecut 35.0 ngkpt 4 4 4 nshiftk 1 shiftk 0 0 0 ############################################## #### SECTION: EPH ############################################## optdriver 7 # Enter EPH driver eph_task 4 # Compute e-ph self-energy (real + imag) tmesh 0 100 4 # [start, step, num_T] in K getddb_filepath "teph4zpr_1_DDB" ddb_ngqpt 4 4 4 # The code expects to find in the DDB # all the IBZ q-points corresponding to a 4x4x4 q-mesh getdvdb_filepath "teph4zpr_2_DVDB" getwfk_filepath "teph4zpr_3o_DS1_WFK" # 4x4x4 k-mesh with 70 bands ############################################## #### SECTION: Sigma_nk ############################################## nkptgw 1 kptgw 0.0 0.0 0.0 bdgw 8 9 zcut 0.01 eV # Activate computation of phonon band structure #ph_prtbands 0 ph_ndivsm 10 ph_nqpath 12 ph_qpath 0.0 0.0 0.0 0.5 0.0 0.5 0.5 0.25 0.75 0.375 0.375 0.75 0.0 0.0 0.0 0.5 0.5 0.5 0.625 0.25 0.625 0.5 0.25 0.75 0.5 0.5 0.5 0.375 0.375 0.75 0.625 0.25 0.625 0.5 0.0 0.5 # Phonon DOS with tetrahedron method #ph_prtdos 0; ph_intmeth 2; ph_wstep 0.0001 eV ph_ngqpt 16 16 16 # Read crystal structure from DEN.nc file structure "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" pp_dirpath "$ABI_PSPDIR" pseudos "O.psp8, Mg-sp-gw.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_4.abo, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 10 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = ZPR computation at the Gamma point for MgO. #%%<END TEST_INFO>
Tip
To run the examples in parallel with e.g 2 MPI processes use:
mpirun -n 2 abinit teph4zpr_4.abi > teph4zpr_4.log 2> err &
The EPH code will automatically distribute the workload using a predefined distribution scheme (not necessarily the most efficient in terms of memory and wall-time). In the last part of the tutorial, we explain how to specify a particular MPI distribution scheme with the eph_np_pqbks input variable.
Let’s now discuss the meaning of the different variables in more detail. We use optdriver 7 to enter the EPH code while eph_task 4 activates the computation of the full self-energy (real + imaginary parts). The paths to the external files (DDB, WFK, DVDB) are specified with the three variables:
This is an excerpt of the input file:
getddb_filepath "teph4zpr_1_DDB" ddb_ngqpt 4 4 4 # The code expects to find in the DDB # all the IBZ q-points corresponding to a 4x4x4 q-mesh getdvdb_filepath "teph4zpr_2_DVDB" getwfk_filepath "teph4zpr_3o_WFK" # 4x4x4 k-mesh with 70 bands
The mesh for electrons (ngkpt, nshiftk and shiftk) must correspond to the one used for the input WFK file. ddb_ngqpt is set to 4x4x4 as this is the \qq-mesh used in the DFPT part to generate the DDB and DVDB files, but the integration in \qq-space is performed with the eph_ngqpt_fine mesh. As eph_ngqpt_fine differs from ddb_ngqpt, the code will automatically activate the interpolation of the DFPT potentials as discussed in the introduction to the EPH code. The \qq-space integration is defined by eph_intmeth and zcut.
We can now have a look at the main output file:
.Version 9.3.1 of ABINIT
.(MPI version, prepared for a x86_64_linux_gnu9.3 computer)
.Copyright (C) 1998-2021 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Tue 10 Nov 2020.
- ( at 22h20 )
- input file -> /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/TestBot_MPI1/tutorespfn_teph4zpr_1-teph4zpr_2-teph4zpr_3-teph4zpr_4-teph4zpr_5-teph4zpr_6-teph4zpr_7-teph4zpr_8/teph4zpr_4.in
- output file -> teph4zpr_4.out
- root for input files -> teph4zpr_4i
- root for output files -> teph4zpr_4o
Symmetries : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need of the present run
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 32 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 30 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 32768 nkpt = 8
================================================================================
P This job should need less than 16.501 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 4.777 Mbytes ; DEN or POT disk file : 0.252 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =312 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_task 4
- fftalg 312
istwfk 2 0 3 0 0 0 7 0
ixc 11
kpt 0.00000000E+00 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 2.50000000E-01
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.27305763E+01
P mkmem 8
natom 2
nband 30
ngfft 32 32 32
nkpt 8
nkptgw 1
nsym 48
ntypat 2
occ 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
optdriver 7
ph_ndivsm 10
ph_ngqpt 16 16 16
ph_nqpath 12
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
wtk 0.01563 0.12500 0.06250 0.09375 0.37500 0.18750
0.04688 0.09375
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
chkinp: Checking input parameters for consistency.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 2, nkpt: 8, mband: 30, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
Average speed of sound partial sums: 0.3110268892E-02 (at units)
- = 6.8043 [km/s]
Debye frequency from partial sums: 0.2394617452E-02 (Ha)
- = 0.1575582594E+02 (THz)
-Debye temperature from partial sums: 0.7561594877E+03 (K)
- Writing phonon DOS to netcdf file: teph4zpr_4o_PHDOS.nc
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- O ONCVPSP r_core= 1.36 1.46 1.26
- 8.00000 6.00000 151103 znucl, zion, pspdat
8 11 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 4.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 1
extension_switch 1
pspatm : epsatm= 6.19401560
--- l ekb(1:nproj) -->
0 5.257212 0.704241
1 -5.135443 -1.451781
2 -4.371486
pspatm: atomic psp has been read and splines computed
- pspini: atom type 2 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- Mg ONCVPSP r_core= 1.16 1.56
- 12.00000 10.00000 150902 znucl, zion, pspdat
8 11 1 4 400 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
3.99000000000000 0.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2
extension_switch 1
pspatm : epsatm= 9.22508734
--- l ekb(1:nproj) -->
0 2.568049 -0.805950
1 -6.605901 -2.664074
pspatm: atomic psp has been read and splines computed
2.46705647E+02 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 30
From bsum_start: 1 to bsum_stop: 30
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 30
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.563 0.073 0.089 -0.002 0.820 -0.301 0.390 0.000 0.000
7 4.490 4.563 0.073 0.089 -0.002 0.820 -0.301 0.390 0.000 0.000
8 4.490 4.563 0.073 0.089 -0.002 0.820 -0.301 0.390 0.000 0.000
9 8.969 8.886 -0.083 -0.085 -0.000 0.981 0.117 -0.201 4.479 4.323
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.323 (OTMS: 4.305)
QP_gap - KS_gap: -0.156 (OTMS: -0.174)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.563 0.073 0.089 -0.002 0.818 -0.310 0.399 0.000 0.000
7 4.490 4.563 0.073 0.089 -0.002 0.818 -0.310 0.399 0.000 0.000
8 4.490 4.563 0.073 0.089 -0.002 0.818 -0.310 0.399 0.000 0.000
9 8.969 8.885 -0.084 -0.086 -0.000 0.981 0.125 -0.210 4.479 4.322
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.322 (OTMS: 4.304)
QP_gap - KS_gap: -0.157 (OTMS: -0.175)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.565 0.075 0.093 -0.002 0.806 -0.369 0.462 0.000 0.000
7 4.490 4.565 0.075 0.093 -0.002 0.806 -0.369 0.462 0.000 0.000
8 4.490 4.565 0.075 0.093 -0.002 0.806 -0.369 0.462 0.000 0.000
9 8.969 8.876 -0.093 -0.095 -0.000 0.978 0.168 -0.263 4.479 4.311
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.311 (OTMS: 4.291)
QP_gap - KS_gap: -0.168 (OTMS: -0.188)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.572 0.082 0.105 -0.003 0.779 -0.462 0.568 0.000 0.000
7 4.490 4.572 0.082 0.105 -0.003 0.779 -0.462 0.568 0.000 0.000
8 4.490 4.572 0.082 0.105 -0.003 0.779 -0.462 0.568 0.000 0.000
9 8.969 8.860 -0.109 -0.112 -0.000 0.974 0.227 -0.340 4.479 4.288
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.288 (OTMS: 4.262)
QP_gap - KS_gap: -0.191 (OTMS: -0.217)
============================================================================================
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_task 4
etotal 0.0000000000E+00
fcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
- fftalg 312
ixc 11
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.27305763E+01
P mkmem 8
natom 2
nband 30
ngfft 32 32 32
nkpt 8
nkptgw 1
nsym 48
ntypat 2
occ 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
optdriver 7
ph_ndivsm 10
ph_ngqpt 16 16 16
ph_nqpath 12
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
strten 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [2] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [3] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [4] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- And optionally:
-
- [5] ABINIT: First-principles approach of materials and nanosystem properties.
- Computer Phys. Comm. 180, 2582-2615 (2009).
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval,
- D. Caliste, R. Caracas, M. Cote, T. Deutsch, L. Genovese, Ph. Ghosez, M. Giantomassi
- S. Goedecker, D.R. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet,
- M.J.T. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf,
- M. Torrent, M.J. Verstraete, G. Zerah, J.W. Zwanziger
- Comment: the third generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT_CPC_v10.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2009
-
- Proc. 0 individual time (sec): cpu= 3.2 wall= 3.2
================================================================================
Calculation completed.
.Delivered 1 WARNINGs and 2 COMMENTs to log file.
+Overall time at end (sec) : cpu= 3.2 wall= 3.2
First of all, we find a section that summarizes the most important parameters:
Number of bands in e-ph self-energy sum: 30
From bsum_start: 1 to bsum_stop: 30
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 1 From: 0.000000E+00 to 0.000000E+00 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [8, 8, 8]
Number of q-points in the IBZ: 29
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of K-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
Note
Note how the acoustic sum-rule (asr), the charge neutrality of the Born effective charges (chneut), the treatment of dipole-dipole interaction in the dynamical matrix (dipdip), are activated by default in v9.
Then we find another section related to MPI parallelism. In this case we are running in sequential but the output will change if we run in paralle (see also eph_np_pqbks). The final message informs the user that the EPH code will either read the qpts from file (if the DVDB contains all of them, in case eph_ngqpt_fine is not defined in the input) or interpolate the scattering potentials from ddb_ngqpt to eph_ngqpt_fine.
=== MPI parallelism === P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 30 P Number of CPUs for parallelism over perturbations: 1 P Number of perturbations treated by this CPU: 6 P Number of CPUs for parallelism over q-points: 1 P Number of q-points in the IBZ treated by this proc: 29 of 29 P Number of CPUs for parallelism over bands: 1 P Number of CPUs for parallelism over spins: 1 P Number of CPUs for parallelism over k-points: 1 P Number of k-point in Sigma_nk treated by this proc: 1 of 1
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
or
Cannot find eph_ngqpt_fine q-points in DVDB --> Activating Fourier interpolation.
Finally, we have the section with the QP results for each spin, \kk-point and temperature, followed by the value of the direct gap computed with the two approaches (linearized QP equation and OTMS):
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.563 0.073 0.089 -0.002 0.820 -0.301 0.390 0.000 0.000
7 4.490 4.563 0.073 0.089 -0.002 0.820 -0.301 0.390 0.000 0.000
8 4.490 4.563 0.073 0.089 -0.002 0.820 -0.301 0.390 0.000 0.000
9 8.969 8.886 -0.083 -0.085 -0.000 0.981 0.117 -0.201 4.479 4.323
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.323 (OTMS: 4.305)
QP_gap - KS_gap: -0.156 (OTMS: -0.174)
The calculation has produced the following output files:
$ ls teph4zpr_4o_* teph4zpr_4o_EBANDS.agr teph4zpr_4o_PHBANDS.agr teph4zpr_4o_PHDOS.nc teph4zpr_4o_PHBST.nc teph4zpr_4o_SIGEPH.nc
where:
- EBANDS.agr → Electron bands in xmgrace format. See also prtebands
- PHBST.agr → Phonon bands in xmgrace format. See also prtphbands ph_ndivsm, ph_nqpath, and ph_qpath.
- PHDOS.nc → Phonon DOS in netcdf format (see prtphdos is given by ph_ngqpt).
- PHPBST.nc → Phonon band structure in netcdf format
- SIGEPH.nc → Netcdf file with \Sigma^{\text{e-ph}} results.
All the QP results are stored in the SIGPEPH.nc netcdf file for all \kk-points, spins and temperatures.
As usual, one can use abiopen with the -p option (--print) to print a summary to terminal:
$ abiopen.py teph4zpr_4o_SIGEPH.nc -p
============================ SigmaEPh calculation ============================
Calculation type: Real + Imaginary part of SigmaEPh
Number of k-points in Sigma_{nk}: 1
sigma_ngkpt: [0 0 0], sigma_erange: [0. 0.]
Max bstart: 5, min bstop: 9
Initial ab-initio q-mesh:
ngqpt: [4 4 4], with nqibz: 8
q-mesh for self-energy integration (eph_ngqpt_fine): [4 4 4]
k-mesh for electrons:
mpdivs: [4 4 4] with shifts [0. 0. 0.] and kptopt: 1
Number of bands included in e-ph self-energy sum: 30
zcut: 0.00037 (Ha), 0.010 (eV)
Number of temperatures: 4, from 0.0 to 300.0 (K)
symsigma: 1
Has Eliashberg function: False
Has Spectral function: False
Printing QP results for 2 temperatures. Use --verbose to print all results.
KS, QP (Z factor) and on-the-mass-shell (OTMS) direct gaps in eV for T = 0.0 K:
Spin k-point KS_gap QPZ_gap QPZ - KS OTMS_gap OTMS - KS
------ --------------------------------- -------- --------- ---------- ---------- -----------
0 [+0.000, +0.000, +0.000] $\Gamma$ 4.479 4.323 -0.156 4.305 -0.174
KS, QP (Z factor) and on-the-mass-shell (OTMS) direct gaps in eV for T = 300.0 K:
Spin k-point KS_gap QPZ_gap QPZ - KS OTMS_gap OTMS - KS
------ --------------------------------- -------- --------- ---------- ---------- -----------
0 [+0.000, +0.000, +0.000] $\Gamma$ 4.479 4.288 -0.191 4.262 -0.217
where QPZ stands for the results obtained with the linearized QP equation and
OTMS are the results with the on-the-mass-shell approach.
We can also plot the most important results by just replacing -p with -e (--expose):
$ abiopen.py teph4zpr_4o_SIGEPH.nc -e
that produces:
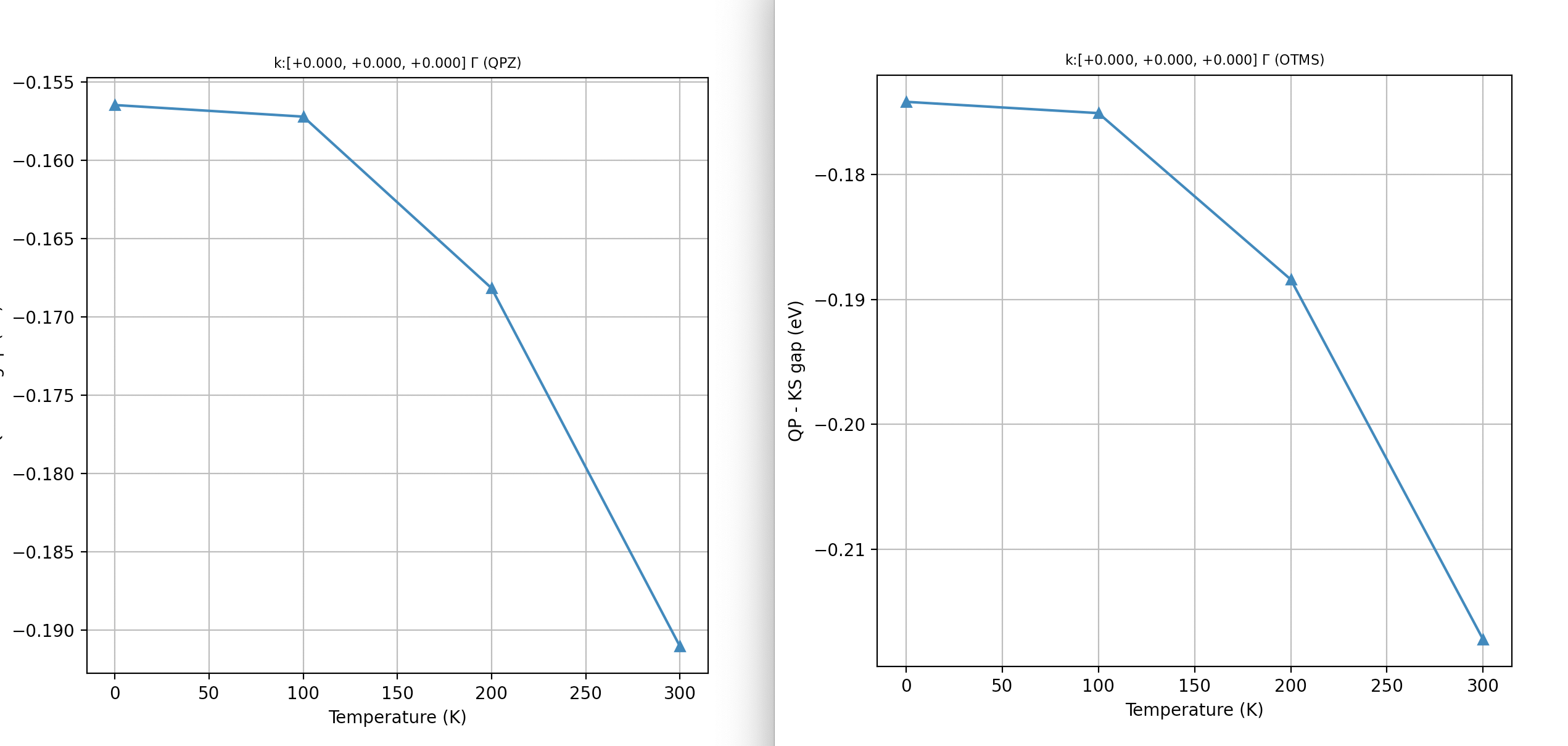
On the left, we have the QP direct gap at \Gamma ad a function of T while the OTMS results are shown in the right panel. In both cases, the QP direct band gap decreases with increasing temperature (remember that MgO is a direct gap semiconductor). This is the so-called Varshni’s effect that is observed in many (but not all) semiconductors.
The ZPR computed with the two approaches differ by ~26 meV but this is expected as the linearized QP equations reduces to the OTMS only if the Z factor is very close to one or if there is some sort of fortuitous cancellation between the CBM/VBM corrections. In our calculation, the Z factor for the VBM is 0.644 while for the CBM we obtain Z = 0.963. These values are reasonable, still it’s not so uncommon to obtain unphysical Z factors in e-ph calculations i.e. values > 1, especially for states far from the band edge as the e-ph self-energy has a lot of structure in frequency-space and the linearized QP approach is not always justified. For this reason, in the rest of the tutorial, we will be focusing on the analysis of the OTMS results.
Important
To compute the imaginary part of \Sigma^{\text{e-ph}} at the KS energy, we strongly recommend to use eph_task -4 as this option activates several important optimizations that are not possible when the full self-energy is wanted. Note, however, that eph_task -4 is not able to provide the full frequency dependence, only the value of the imaginary part at the KS eigenvalue. The computation of spectral functions and Eliashberg functions therefore requires eph_task +4.
Convergence w.r.t. nband¶
At this point it should not be so difficult to write an input file to perform ZPR calculations for different values of nband for a fixed \qq-mesh. It is just a matter of adding the following set of variables to the previous input file:
ndtset 3 nband: 20 nband+ 20
An example is given in
# ZPR computation at Gamma for MgO. Convergence of ZPR wrt nband ndtset 3 nband: 20 nband+ 20 ecut 35.0 ngkpt 4 4 4 nshiftk 1 shiftk 0 0 0 ############################################## #### SECTION: eph ############################################## optdriver 7 eph_task 4 tmesh 0 100 4 # [start, step, num_T] in K getddb_filepath "teph4zpr_1_DDB" ddb_ngqpt 4 4 4 getdvdb_filepath "teph4zpr_2_DVDB" getwfk_filepath "teph4zpr_3o_DS1_WFK" ############################################## #### SECTION: Sigma_nk ############################################## nkptgw 1 kptgw 0.0 0.0 0.0 bdgw 8 9 zcut 0.01 eV structure = "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" pp_dirpath = "$ABI_PSPDIR" pseudos = "O.psp8, Mg-sp-gw.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_5.abo, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 10 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = ZPR computation at Gamma for MgO. Convergence of ZPR wrt nband #%%<END TEST_INFO>
Run the calculation, as usual, using
abinit teph4zpr_5.abi > teph4zpr_5.log 2> err &
Now we can extract the results from the output file
.Version 9.3.1 of ABINIT
.(MPI version, prepared for a x86_64_linux_gnu9.3 computer)
.Copyright (C) 1998-2021 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Tue 10 Nov 2020.
- ( at 22h20 )
- input file -> /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/TestBot_MPI1/tutorespfn_teph4zpr_1-teph4zpr_2-teph4zpr_3-teph4zpr_4-teph4zpr_5-teph4zpr_6-teph4zpr_7-teph4zpr_8/teph4zpr_5.in
- output file -> teph4zpr_5.out
- root for input files -> teph4zpr_5i
- root for output files -> teph4zpr_5o
DATASET 1 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 32 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 20 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 32768 nkpt = 8
================================================================================
P This job should need less than 14.907 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 3.186 Mbytes ; DEN or POT disk file : 0.252 Mbytes.
================================================================================
DATASET 2 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 32 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 40 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 32768 nkpt = 8
================================================================================
P This job should need less than 18.095 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 6.369 Mbytes ; DEN or POT disk file : 0.252 Mbytes.
================================================================================
DATASET 3 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 3.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 32 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 60 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 32768 nkpt = 8
================================================================================
P This job should need less than 21.282 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 9.553 Mbytes ; DEN or POT disk file : 0.252 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =312 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_task 4
- fftalg 312
istwfk 2 0 3 0 0 0 7 0
ixc 11
jdtset 1 2 3
kpt 0.00000000E+00 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 2.50000000E-01
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.27305763E+01
P mkmem 8
natom 2
nband1 20
nband2 40
nband3 60
ndtset 3
ngfft 32 32 32
nkpt 8
nkptgw 1
nsym 48
ntypat 2
occ1 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
optdriver 7
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
wtk 0.01563 0.12500 0.06250 0.09375 0.37500 0.18750
0.04688 0.09375
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
chkinp: Checking input parameters for consistency, jdtset= 3.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 2, nkpt: 8, mband: 20, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
Average speed of sound partial sums: 0.3115170319E-02 (at units)
- = 6.8150 [km/s]
Debye frequency from partial sums: 0.2398391094E-02 (Ha)
- = 0.1578065532E+02 (THz)
-Debye temperature from partial sums: 0.7573511083E+03 (K)
- Writing phonon DOS to netcdf file: teph4zpr_5o_DS1_PHDOS.nc
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- O ONCVPSP r_core= 1.36 1.46 1.26
- 8.00000 6.00000 151103 znucl, zion, pspdat
8 11 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 4.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 1
extension_switch 1
pspatm : epsatm= 6.19401560
--- l ekb(1:nproj) -->
0 5.257212 0.704241
1 -5.135443 -1.451781
2 -4.371486
pspatm: atomic psp has been read and splines computed
- pspini: atom type 2 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- Mg ONCVPSP r_core= 1.16 1.56
- 12.00000 10.00000 150902 znucl, zion, pspdat
8 11 1 4 400 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
3.99000000000000 0.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2
extension_switch 1
pspatm : epsatm= 9.22508734
--- l ekb(1:nproj) -->
0 2.568049 -0.805950
1 -6.605901 -2.664074
pspatm: atomic psp has been read and splines computed
2.46705647E+02 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 20
From bsum_start: 1 to bsum_stop: 20
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 20
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.548 0.058 0.071 -0.002 0.823 -0.077 0.147 0.000 0.000
7 4.490 4.548 0.058 0.071 -0.002 0.823 -0.077 0.147 0.000 0.000
8 4.490 4.548 0.058 0.071 -0.002 0.823 -0.077 0.147 0.000 0.000
9 8.969 8.892 -0.077 -0.079 -0.000 0.981 0.132 -0.211 4.479 4.344
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.344 (OTMS: 4.330)
QP_gap - KS_gap: -0.135 (OTMS: -0.149)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.548 0.058 0.071 -0.002 0.822 -0.080 0.151 0.000 0.000
7 4.490 4.548 0.058 0.071 -0.002 0.822 -0.080 0.151 0.000 0.000
8 4.490 4.548 0.058 0.071 -0.002 0.822 -0.080 0.151 0.000 0.000
9 8.969 8.891 -0.078 -0.079 -0.000 0.981 0.141 -0.220 4.479 4.343
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.343 (OTMS: 4.329)
QP_gap - KS_gap: -0.136 (OTMS: -0.150)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.549 0.059 0.073 -0.002 0.809 -0.100 0.173 0.000 0.000
7 4.490 4.549 0.059 0.073 -0.002 0.809 -0.100 0.173 0.000 0.000
8 4.490 4.549 0.059 0.073 -0.002 0.809 -0.100 0.173 0.000 0.000
9 8.969 8.883 -0.085 -0.087 -0.000 0.979 0.187 -0.275 4.479 4.335
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.335 (OTMS: 4.319)
QP_gap - KS_gap: -0.144 (OTMS: -0.160)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.553 0.063 0.080 -0.003 0.783 -0.130 0.211 0.000 0.000
7 4.490 4.553 0.063 0.080 -0.003 0.783 -0.130 0.211 0.000 0.000
8 4.490 4.553 0.063 0.080 -0.003 0.783 -0.130 0.211 0.000 0.000
9 8.969 8.869 -0.099 -0.102 -0.000 0.974 0.252 -0.354 4.479 4.317
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.317 (OTMS: 4.296)
QP_gap - KS_gap: -0.162 (OTMS: -0.183)
============================================================================================
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 2, nkpt: 8, mband: 40, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
Average speed of sound partial sums: 0.3115170319E-02 (at units)
- = 6.8150 [km/s]
Debye frequency from partial sums: 0.2398391094E-02 (Ha)
- = 0.1578065532E+02 (THz)
-Debye temperature from partial sums: 0.7573511083E+03 (K)
- Writing phonon DOS to netcdf file: teph4zpr_5o_DS2_PHDOS.nc
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 40
From bsum_start: 1 to bsum_stop: 40
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 40
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.652 0.162 0.199 -0.002 0.817 -0.592 0.791 0.000 0.000
7 4.490 4.652 0.162 0.199 -0.002 0.817 -0.592 0.791 0.000 0.000
8 4.490 4.652 0.162 0.199 -0.002 0.817 -0.592 0.791 0.000 0.000
9 8.969 8.904 -0.064 -0.066 -0.000 0.981 0.096 -0.162 4.479 4.252
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.252 (OTMS: 4.215)
QP_gap - KS_gap: -0.227 (OTMS: -0.264)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.653 0.163 0.200 -0.002 0.815 -0.607 0.807 0.000 0.000
7 4.490 4.653 0.163 0.200 -0.002 0.815 -0.607 0.807 0.000 0.000
8 4.490 4.653 0.163 0.200 -0.002 0.815 -0.607 0.807 0.000 0.000
9 8.969 8.904 -0.065 -0.066 -0.000 0.980 0.104 -0.170 4.479 4.251
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.251 (OTMS: 4.213)
QP_gap - KS_gap: -0.228 (OTMS: -0.266)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.663 0.173 0.216 -0.002 0.802 -0.711 0.927 0.000 0.000
7 4.490 4.663 0.173 0.216 -0.002 0.802 -0.711 0.927 0.000 0.000
8 4.490 4.663 0.173 0.216 -0.002 0.802 -0.711 0.927 0.000 0.000
9 8.969 8.899 -0.069 -0.071 -0.000 0.978 0.144 -0.215 4.479 4.237
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.237 (OTMS: 4.192)
QP_gap - KS_gap: -0.242 (OTMS: -0.287)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.683 0.193 0.249 -0.003 0.775 -0.882 1.131 0.000 0.000
7 4.490 4.683 0.193 0.249 -0.003 0.775 -0.882 1.131 0.000 0.000
8 4.490 4.683 0.193 0.249 -0.003 0.775 -0.882 1.131 0.000 0.000
9 8.969 8.889 -0.079 -0.082 -0.000 0.973 0.198 -0.279 4.479 4.207
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.207 (OTMS: 4.148)
QP_gap - KS_gap: -0.272 (OTMS: -0.331)
============================================================================================
================================================================================
== DATASET 3 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 3, }
dimensions: {natom: 2, nkpt: 8, mband: 60, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
Average speed of sound partial sums: 0.3115170319E-02 (at units)
- = 6.8150 [km/s]
Debye frequency from partial sums: 0.2398391094E-02 (Ha)
- = 0.1578065532E+02 (THz)
-Debye temperature from partial sums: 0.7573511083E+03 (K)
- Writing phonon DOS to netcdf file: teph4zpr_5o_DS3_PHDOS.nc
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 60
From bsum_start: 1 to bsum_stop: 60
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 60
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.499 0.010 0.012 -0.002 0.814 -0.960 0.972 0.000 0.000
7 4.490 4.499 0.010 0.012 -0.002 0.814 -0.960 0.972 0.000 0.000
8 4.490 4.499 0.010 0.012 -0.002 0.814 -0.960 0.972 0.000 0.000
9 8.969 8.880 -0.089 -0.091 -0.000 0.980 0.068 -0.158 4.479 4.381
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.381 (OTMS: 4.377)
QP_gap - KS_gap: -0.098 (OTMS: -0.102)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.498 0.009 0.011 -0.002 0.812 -0.982 0.993 0.000 0.000
7 4.490 4.498 0.009 0.011 -0.002 0.812 -0.982 0.993 0.000 0.000
8 4.490 4.498 0.009 0.011 -0.002 0.812 -0.982 0.993 0.000 0.000
9 8.969 8.879 -0.090 -0.091 -0.000 0.980 0.075 -0.166 4.479 4.381
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.381 (OTMS: 4.377)
QP_gap - KS_gap: -0.098 (OTMS: -0.102)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.493 0.003 0.004 -0.003 0.799 -1.139 1.143 0.000 0.000
7 4.490 4.493 0.003 0.004 -0.003 0.799 -1.139 1.143 0.000 0.000
8 4.490 4.493 0.003 0.004 -0.003 0.799 -1.139 1.143 0.000 0.000
9 8.969 8.870 -0.098 -0.101 -0.000 0.977 0.110 -0.210 4.479 4.378
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.378 (OTMS: 4.375)
QP_gap - KS_gap: -0.101 (OTMS: -0.104)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.486 -0.003 -0.005 -0.003 0.771 -1.401 1.396 0.000 0.000
7 4.490 4.486 -0.003 -0.005 -0.003 0.771 -1.401 1.396 0.000 0.000
8 4.490 4.486 -0.003 -0.005 -0.003 0.771 -1.401 1.396 0.000 0.000
9 8.969 8.854 -0.115 -0.118 -0.000 0.973 0.155 -0.273 4.479 4.367
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.367 (OTMS: 4.365)
QP_gap - KS_gap: -0.112 (OTMS: -0.114)
============================================================================================
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_task 4
etotal1 0.0000000000E+00
etotal2 0.0000000000E+00
etotal3 0.0000000000E+00
fcart1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
- fftalg 312
ixc 11
jdtset 1 2 3
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.27305763E+01
P mkmem 8
natom 2
nband1 20
nband2 40
nband3 60
ndtset 3
ngfft 32 32 32
nkpt 8
nkptgw 1
nsym 48
ntypat 2
occ1 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
optdriver 7
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
strten1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [2] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [3] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [4] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- And optionally:
-
- [5] ABINIT: First-principles approach of materials and nanosystem properties.
- Computer Phys. Comm. 180, 2582-2615 (2009).
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval,
- D. Caliste, R. Caracas, M. Cote, T. Deutsch, L. Genovese, Ph. Ghosez, M. Giantomassi
- S. Goedecker, D.R. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet,
- M.J.T. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf,
- M. Torrent, M.J. Verstraete, G. Zerah, J.W. Zwanziger
- Comment: the third generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT_CPC_v10.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2009
-
- Proc. 0 individual time (sec): cpu= 10.8 wall= 10.8
================================================================================
Calculation completed.
.Delivered 3 WARNINGs and 9 COMMENTs to log file.
+Overall time at end (sec) : cpu= 10.8 wall= 10.8
or, alternatively, use the AbiPy abicomp script to post-process the results stored in the SIGPEPH files:
abicomp.py sigeph teph4zpr_5o_DS*_SIGEPH.nc -e
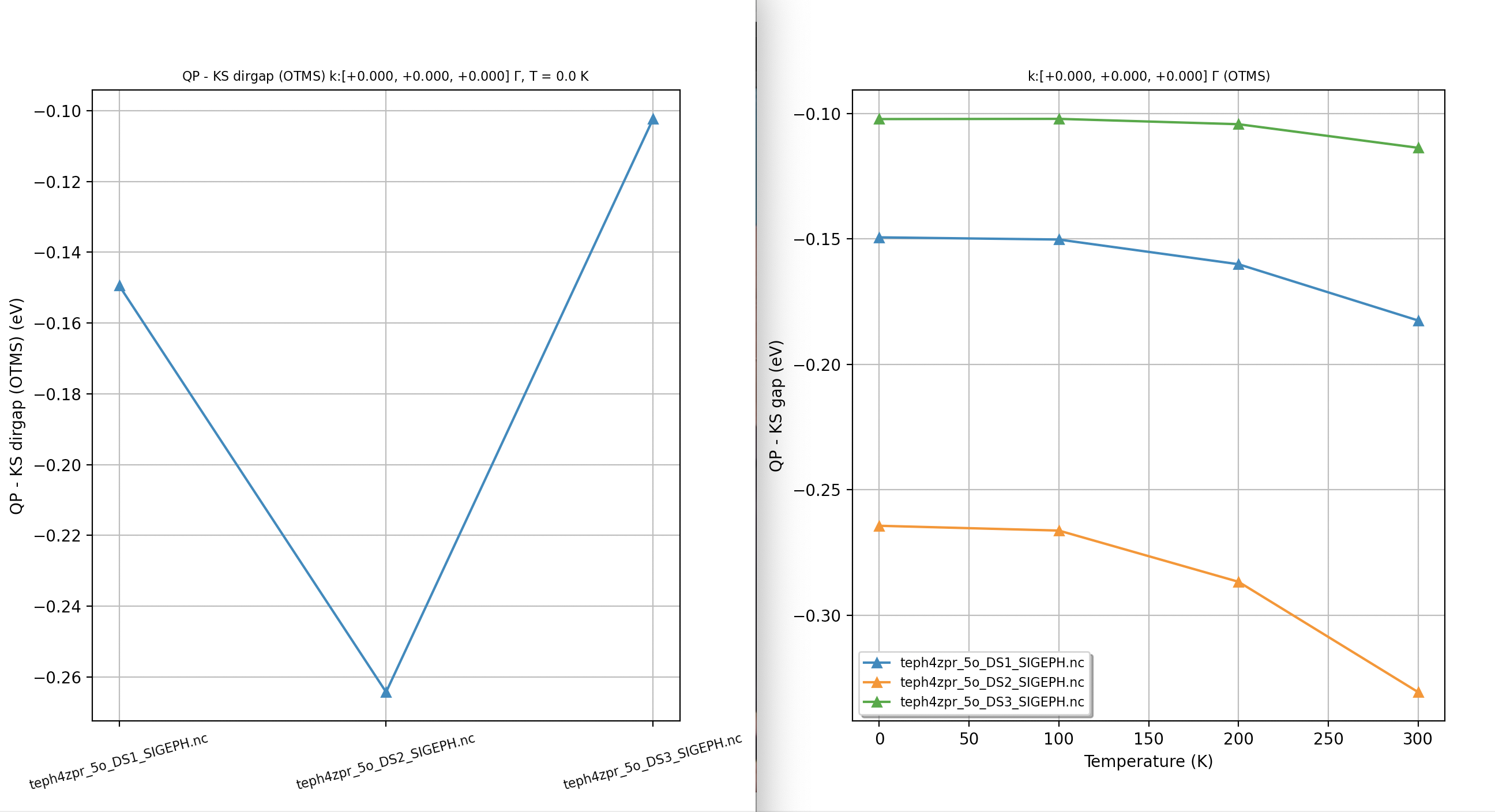
The figure on the left shows the convergence of the OTMS ZPR as a function of nband. The figure on the right, gives the correction to the KS band gas as a function of T obtained for different numbers of bands. A similar behaviour is observed also for the linearized equation.
The results are quite disappointing in the sense that the QP gaps are far from being converged and the convergence rate is even worse if we look at the QP energies (the same behaviour is observed in GW). A more careful (and expensive) convergence study would reveal that 300 bands are needed. This computation, although feasible, would be too costly for a tutorial and is therefore left as an extra exercise. In the next section, we will see that the EPH code provides a much more efficient algorithm to accelerate the convergence.
Exercise
Change the input file to use mixprec 1 and boxcutmin 1.1. Rerun the calculation and compare with the previous results. Do you see significant differences? What about the wall-time and the memory allocated for W(\rr, \RR) Hint: use
grep log <<< MEM
to extract the lines where the most imporant memory allocation are reported.
How to reduce the number of bands with the Sternheimer method¶
To activate the Sternheimer approach, we need to set eph_stern to 1 and use getpot_filepath to specify the external file with the GS KS potential. This file is produced at the end of the GS calculations provided prtpot is set to 1 in the input file.
Important
The Sternheimer equation is solved non-self-consistently using max nline NSCF iterations and the solver stops when the first-order wavefunction is converged within tolwfr. Default values for these two variables are provided if not specified by the user in the input file. The code aborts if the solver cannot converge. This may happen if the first nband states are not well separated from the remaining high-energy states. Increasing nband should solve the problem.
An example of input file is provided in:
# Convergence of ZPR wrt nband with the Sternheimer method ndtset 4 nband: 10 nband+ 10 ecut 35.0 ngkpt 4 4 4 nshiftk 1 shiftk 0 0 0 ############################################## #### SECTION: eph ############################################## optdriver 7 eph_task 4 ddb_ngqpt 4 4 4 tmesh 0 100 4 # [start, step, num_T] in K getddb_filepath "teph4zpr_1_DDB" getdvdb_filepath "teph4zpr_2_DVDB" getwfk_filepath "teph4zpr_3o_DS1_WFK" eph_stern 1 getpot_filepath "MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc" mixprec 1 boxcutmin 1.1 prtphdos 0 # Disable computation of PHDOS to save some time ############################################## #### SECTION: Sigma_nk ############################################## nkptgw 1 kptgw 0.0 0.0 0.0 bdgw 8 9 zcut 0.01 eV structure = "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" pp_dirpath = "$ABI_PSPDIR" pseudos = "O.psp8, Mg-sp-gw.psp8" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_6.abo, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 10 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = Convergence of ZPR wrt nband with the Sternheimer method #%%<END TEST_INFO>
and can be run with:
abinit teph4zpr_6.abi > teph4zpr_6.log 2> err &
Four calculations are performed with different values of nband:
ndtset 4 nband: 10 nband+ 10
in order to monitor the convergence of the QP corrections.
To analyze the convergence behavior, we can extract the results from the main output file
.Version 9.3.1 of ABINIT
.(MPI version, prepared for a x86_64_linux_gnu9.3 computer)
.Copyright (C) 1998-2021 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Tue 10 Nov 2020.
- ( at 22h21 )
- input file -> /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/TestBot_MPI1/tutorespfn_teph4zpr_1-teph4zpr_2-teph4zpr_3-teph4zpr_4-teph4zpr_5-teph4zpr_6-teph4zpr_7-teph4zpr_8/teph4zpr_6.in
- output file -> teph4zpr_6.out
- root for input files -> teph4zpr_6i
- root for output files -> teph4zpr_6o
DATASET 1 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 10 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 5832 nkpt = 8
================================================================================
P This job should need less than 4.799 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 1.594 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
DATASET 2 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 20 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 5832 nkpt = 8
================================================================================
P This job should need less than 6.411 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 3.186 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
DATASET 3 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 3.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 30 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 5832 nkpt = 8
================================================================================
P This job should need less than 8.028 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 4.777 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
DATASET 4 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 4.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 40 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 5832 nkpt = 8
================================================================================
P This job should need less than 9.650 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 6.369 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =312 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
boxcutmin 1.10000000E+00
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_stern 1
eph_task 4
- fftalg 312
istwfk 2 0 3 0 0 0 7 0
ixc 11
jdtset 1 2 3 4
kpt 0.00000000E+00 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 2.50000000E-01
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.27305763E+01
mixprec 1
P mkmem 8
natom 2
nband1 10
nband2 20
nband3 30
nband4 40
ndtset 4
ngfft 18 18 18
nkpt 8
nkptgw 1
nline 100
nsym 48
ntypat 2
occ1 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
occ4 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000
optdriver 7
prtphdos 0
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tolwfr 1.00000000E-16
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
wtk 0.01563 0.12500 0.06250 0.09375 0.37500 0.18750
0.04688 0.09375
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
chkinp: Checking input parameters for consistency, jdtset= 3.
chkinp: Checking input parameters for consistency, jdtset= 4.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 2, nkpt: 8, mband: 10, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- O ONCVPSP r_core= 1.36 1.46 1.26
- 8.00000 6.00000 151103 znucl, zion, pspdat
8 11 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 4.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 1
extension_switch 1
pspatm : epsatm= 6.19401560
--- l ekb(1:nproj) -->
0 5.257212 0.704241
1 -5.135443 -1.451781
2 -4.371486
pspatm: atomic psp has been read and splines computed
- pspini: atom type 2 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- Mg ONCVPSP r_core= 1.16 1.56
- 12.00000 10.00000 150902 znucl, zion, pspdat
8 11 1 4 400 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
3.99000000000000 0.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2
extension_switch 1
pspatm : epsatm= 9.22508734
--- l ekb(1:nproj) -->
0 2.568049 -0.805950
1 -6.605901 -2.664074
pspatm: atomic psp has been read and splines computed
2.46705647E+02 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 10
From bsum_start: 1 to bsum_stop: 10
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 10
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.571 0.081 0.098 -0.002 0.829 -3.998 4.096 0.000 0.000
7 4.490 4.571 0.081 0.098 -0.002 0.829 -3.998 4.096 0.000 0.000
8 4.490 4.571 0.081 0.098 -0.002 0.829 -3.998 4.096 0.000 0.000
9 8.969 8.888 -0.081 -0.082 -0.000 0.983 -0.054 -0.028 4.479 4.317
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.317 (OTMS: 4.299)
QP_gap - KS_gap: -0.162 (OTMS: -0.180)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.571 0.081 0.098 -0.002 0.827 -4.088 4.186 0.000 0.000
7 4.490 4.571 0.081 0.098 -0.002 0.827 -4.088 4.186 0.000 0.000
8 4.490 4.571 0.081 0.098 -0.002 0.827 -4.088 4.186 0.000 0.000
9 8.969 8.887 -0.082 -0.083 -0.000 0.982 -0.050 -0.033 4.479 4.316
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.316 (OTMS: 4.298)
QP_gap - KS_gap: -0.163 (OTMS: -0.181)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.573 0.084 0.103 -0.002 0.816 -4.735 4.837 0.000 0.000
7 4.490 4.573 0.084 0.103 -0.002 0.816 -4.735 4.837 0.000 0.000
8 4.490 4.573 0.084 0.103 -0.002 0.816 -4.735 4.837 0.000 0.000
9 8.969 8.879 -0.089 -0.091 -0.000 0.980 -0.035 -0.056 4.479 4.306
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.306 (OTMS: 4.285)
QP_gap - KS_gap: -0.173 (OTMS: -0.194)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.581 0.091 0.115 -0.003 0.790 -5.812 5.927 0.000 0.000
7 4.490 4.581 0.091 0.115 -0.003 0.790 -5.812 5.927 0.000 0.000
8 4.490 4.581 0.091 0.115 -0.003 0.790 -5.812 5.927 0.000 0.000
9 8.969 8.864 -0.105 -0.107 -0.000 0.976 -0.023 -0.084 4.479 4.284
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.284 (OTMS: 4.257)
QP_gap - KS_gap: -0.195 (OTMS: -0.222)
============================================================================================
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 2, nkpt: 8, mband: 20, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 20
From bsum_start: 1 to bsum_stop: 20
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 20
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.570 0.080 0.097 -0.002 0.823 -3.999 4.096 0.000 0.000
7 4.490 4.570 0.080 0.097 -0.002 0.823 -3.999 4.096 0.000 0.000
8 4.490 4.570 0.080 0.097 -0.002 0.823 -3.999 4.096 0.000 0.000
9 8.969 8.888 -0.081 -0.083 -0.000 0.981 -0.055 -0.028 4.479 4.318
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.318 (OTMS: 4.299)
QP_gap - KS_gap: -0.161 (OTMS: -0.180)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.570 0.080 0.097 -0.002 0.822 -4.089 4.187 0.000 0.000
7 4.490 4.570 0.080 0.097 -0.002 0.822 -4.089 4.187 0.000 0.000
8 4.490 4.570 0.080 0.097 -0.002 0.822 -4.089 4.187 0.000 0.000
9 8.969 8.887 -0.082 -0.083 -0.000 0.981 -0.051 -0.033 4.479 4.317
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.317 (OTMS: 4.298)
QP_gap - KS_gap: -0.162 (OTMS: -0.181)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.572 0.082 0.102 -0.002 0.809 -4.736 4.837 0.000 0.000
7 4.490 4.572 0.082 0.102 -0.002 0.809 -4.736 4.837 0.000 0.000
8 4.490 4.572 0.082 0.102 -0.002 0.809 -4.736 4.837 0.000 0.000
9 8.969 8.879 -0.090 -0.092 -0.000 0.979 -0.036 -0.055 4.479 4.307
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.307 (OTMS: 4.286)
QP_gap - KS_gap: -0.172 (OTMS: -0.193)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.579 0.089 0.114 -0.003 0.783 -5.813 5.927 0.000 0.000
7 4.490 4.579 0.089 0.114 -0.003 0.783 -5.813 5.927 0.000 0.000
8 4.490 4.579 0.089 0.114 -0.003 0.783 -5.813 5.927 0.000 0.000
9 8.969 8.864 -0.105 -0.108 -0.000 0.974 -0.024 -0.083 4.479 4.285
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.285 (OTMS: 4.258)
QP_gap - KS_gap: -0.194 (OTMS: -0.221)
============================================================================================
================================================================================
== DATASET 3 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 3, }
dimensions: {natom: 2, nkpt: 8, mband: 30, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 30
From bsum_start: 1 to bsum_stop: 30
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 30
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.569 0.079 0.097 -0.002 0.820 -4.000 4.097 0.000 0.000
7 4.490 4.569 0.079 0.097 -0.002 0.820 -4.000 4.097 0.000 0.000
8 4.490 4.569 0.079 0.097 -0.002 0.820 -4.000 4.097 0.000 0.000
9 8.969 8.888 -0.081 -0.083 -0.000 0.981 -0.055 -0.028 4.479 4.319
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.319 (OTMS: 4.300)
QP_gap - KS_gap: -0.160 (OTMS: -0.179)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.569 0.079 0.097 -0.002 0.818 -4.091 4.188 0.000 0.000
7 4.490 4.569 0.079 0.097 -0.002 0.818 -4.091 4.188 0.000 0.000
8 4.490 4.569 0.079 0.097 -0.002 0.818 -4.091 4.188 0.000 0.000
9 8.969 8.887 -0.082 -0.083 -0.000 0.981 -0.051 -0.032 4.479 4.318
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.318 (OTMS: 4.298)
QP_gap - KS_gap: -0.161 (OTMS: -0.181)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.571 0.082 0.101 -0.002 0.806 -4.737 4.839 0.000 0.000
7 4.490 4.571 0.082 0.101 -0.002 0.806 -4.737 4.839 0.000 0.000
8 4.490 4.571 0.082 0.101 -0.002 0.806 -4.737 4.839 0.000 0.000
9 8.969 8.879 -0.090 -0.092 -0.000 0.978 -0.037 -0.055 4.479 4.308
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.308 (OTMS: 4.286)
QP_gap - KS_gap: -0.171 (OTMS: -0.193)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.578 0.088 0.113 -0.003 0.779 -5.815 5.928 0.000 0.000
7 4.490 4.578 0.088 0.113 -0.003 0.779 -5.815 5.928 0.000 0.000
8 4.490 4.578 0.088 0.113 -0.003 0.779 -5.815 5.928 0.000 0.000
9 8.969 8.864 -0.105 -0.108 -0.000 0.974 -0.025 -0.083 4.479 4.286
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.286 (OTMS: 4.258)
QP_gap - KS_gap: -0.193 (OTMS: -0.221)
============================================================================================
================================================================================
== DATASET 4 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 4, }
dimensions: {natom: 2, nkpt: 8, mband: 40, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 40
From bsum_start: 1 to bsum_stop: 40
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 40
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.570 0.080 0.098 -0.002 0.817 -4.002 4.100 0.000 0.000
7 4.490 4.570 0.080 0.098 -0.002 0.817 -4.002 4.100 0.000 0.000
8 4.490 4.570 0.080 0.098 -0.002 0.817 -4.002 4.100 0.000 0.000
9 8.969 8.888 -0.081 -0.082 -0.000 0.981 -0.055 -0.027 4.479 4.318
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.318 (OTMS: 4.298)
QP_gap - KS_gap: -0.161 (OTMS: -0.181)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.570 0.080 0.098 -0.002 0.815 -4.092 4.191 0.000 0.000
7 4.490 4.570 0.080 0.098 -0.002 0.815 -4.092 4.191 0.000 0.000
8 4.490 4.570 0.080 0.098 -0.002 0.815 -4.092 4.191 0.000 0.000
9 8.969 8.887 -0.082 -0.083 -0.000 0.980 -0.051 -0.032 4.479 4.317
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.317 (OTMS: 4.297)
QP_gap - KS_gap: -0.162 (OTMS: -0.182)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.572 0.082 0.103 -0.002 0.802 -4.739 4.842 0.000 0.000
7 4.490 4.572 0.082 0.103 -0.002 0.802 -4.739 4.842 0.000 0.000
8 4.490 4.572 0.082 0.103 -0.002 0.802 -4.739 4.842 0.000 0.000
9 8.969 8.879 -0.090 -0.092 -0.000 0.978 -0.037 -0.055 4.479 4.307
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.307 (OTMS: 4.285)
QP_gap - KS_gap: -0.172 (OTMS: -0.194)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.579 0.089 0.115 -0.003 0.775 -5.817 5.932 0.000 0.000
7 4.490 4.579 0.089 0.115 -0.003 0.775 -5.817 5.932 0.000 0.000
8 4.490 4.579 0.089 0.115 -0.003 0.775 -5.817 5.932 0.000 0.000
9 8.969 8.864 -0.105 -0.108 -0.000 0.973 -0.025 -0.082 4.479 4.285
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.285 (OTMS: 4.256)
QP_gap - KS_gap: -0.194 (OTMS: -0.223)
============================================================================================
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
boxcutmin 1.10000000E+00
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_stern 1
eph_task 4
etotal1 0.0000000000E+00
etotal2 0.0000000000E+00
etotal3 0.0000000000E+00
etotal4 0.0000000000E+00
fcart1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart4 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
- fftalg 312
ixc 11
jdtset 1 2 3 4
kptrlatt 4 0 0 0 4 0 0 0 4
kptrlen 2.27305763E+01
mixprec 1
P mkmem 8
natom 2
nband1 10
nband2 20
nband3 30
nband4 40
ndtset 4
ngfft 18 18 18
nkpt 8
nkptgw 1
nline 100
nsym 48
ntypat 2
occ1 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
occ4 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000
optdriver 7
prtphdos 0
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
strten1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten4 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tolwfr 1.00000000E-16
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [2] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [3] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [4] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- And optionally:
-
- [5] ABINIT: First-principles approach of materials and nanosystem properties.
- Computer Phys. Comm. 180, 2582-2615 (2009).
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval,
- D. Caliste, R. Caracas, M. Cote, T. Deutsch, L. Genovese, Ph. Ghosez, M. Giantomassi
- S. Goedecker, D.R. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet,
- M.J.T. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf,
- M. Torrent, M.J. Verstraete, G. Zerah, J.W. Zwanziger
- Comment: the third generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT_CPC_v10.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2009
-
- Proc. 0 individual time (sec): cpu= 15.7 wall= 15.9
================================================================================
Calculation completed.
.Delivered 8 WARNINGs and 17 COMMENTs to log file.
+Overall time at end (sec) : cpu= 15.7 wall= 15.9
or, alternatively, we can pass the list of SIGEPH files to the abicomp script and use the
sigpeh command:
abicomp.py sigeph teph4zpr_6o_DS*_SIGEPH.nc -e
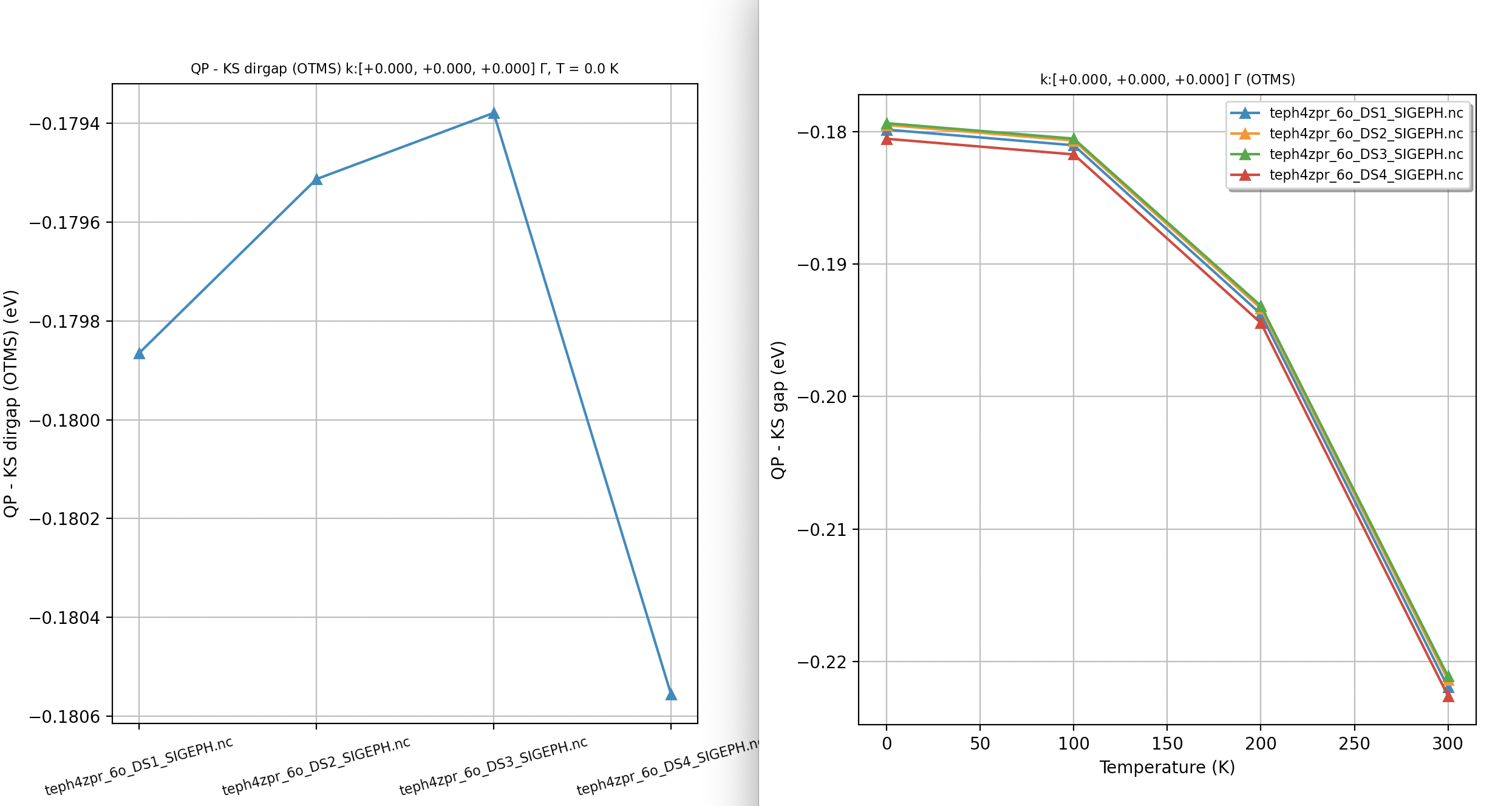
Now the convergence is much better. Note the different scale on the y-axis.
Important
The big advange of the Sternheimer method is that we don’t need to compute WFK files with many empty bands in order to converge the real part of the self-energy. This means that one can use the computing power to densify the \kk-mesh while keeping the number of empty states at a reasonable level. Producing a WFK file with 1000 bands and a 100^3 \kk-mesh is indeed way more expensive than performing the same computation with, let’s say, 50 bands.
Note, however, that the cost of the Sternheimer method quickly increases with nband due to the orthogonalization process. This means that a ZPR calculation with 300 bands (occ + empty) without the Sternheimer method is much faster than the same computation done with eph_stern 1. As a matter of fact, we use the Sternheimer method so that we don’t need 300 bands to converge the results.
Exercise
Generate a new WFK with nband ~ %mpw and run a ZPR calculation without the Sternheimer method. Compare the sum-over-states results with the Sternheimer ones. Why it is not a good idea to use nband >= mpw?
Convergence of the ZPR w.r.t. the q-mesh¶
Now we fix this value of nband to 20 and perform a convergence study for the \qq-sampling. Unfortunately, we won’t be able to fully convergence the results but, at least, you will get an idea on how to perform this kind of convergence study.
In the input teph4zpr_3.abi, we have computed WFK files on different \kk-meshes and a relatively small number of empty states (25 - 5 = 20). We can finally use these WFK files to perform ZPR calculations by just adding:
ndtset 3 nband 20 ngkpt1 4 4 4 eph_ngqpt_fine1 4 4 4 getwfk_filepath1 "teph4zpr_3o_DS1_WFK" ngkpt2 8 8 8 eph_ngqpt_fine2 8 8 8 getwfk_filepath2 "teph4zpr_3o_DS2_WFK" ngkpt3 12 12 12 eph_ngqpt_fine3 12 12 12 getwfk_filepath3 "teph4zpr_3o_DS3_WFK" eph_stern 1 getpot_filepath "MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc"
# Convergence of ZPR wrt q-sampling with Sternheimer method ndtset 3 nband 20 ngkpt1 4 4 4 eph_ngqpt_fine1 4 4 4 getwfk_filepath1 "teph4zpr_3o_DS1_WFK" ngkpt2 8 8 8 eph_ngqpt_fine2 8 8 8 getwfk_filepath2 "teph4zpr_3o_DS2_WFK" ngkpt3 12 12 12 eph_ngqpt_fine3 12 12 12 getwfk_filepath3 "teph4zpr_3o_DS3_WFK" eph_stern 1 getpot_filepath "MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc" # nline tolwfr getddb_filepath "teph4zpr_1_DDB" ddb_ngqpt 4 4 4 getdvdb_filepath "teph4zpr_2_DVDB" ecut 35.0 nshiftk 1 shiftk 0 0 0 mixprec 1 boxcutmin 1.1 # Important to speedup the calculation and reduce memory. ############################################## #### SECTION: eph ############################################## optdriver 7 eph_task 4 tmesh 0 100 4 # [start, step, num_T] in K prtphdos 0 # Disable computation of PHDOS to save some time ############################################## #### SECTION: Sigma_nk ############################################## nkptgw 1 kptgw 0.0 0.0 0.0 bdgw 8 9 zcut 0.01 eV pp_dirpath = "$ABI_PSPDIR" pseudos = "O.psp8, Mg-sp-gw.psp8" structure = "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_7.abo, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 10 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = Convergence of ZPR wrt q-sampling with Sternheimer method #%%<END TEST_INFO>
Run the calculation, as usual, with:
abinit teph4zpr_7.abi > teph4zpr_7.log 2> err &
The output file is reported here for your convenience:
.Version 9.3.1 of ABINIT
.(MPI version, prepared for a x86_64_linux_gnu9.3 computer)
.Copyright (C) 1998-2021 ABINIT group .
ABINIT comes with ABSOLUTELY NO WARRANTY.
It is free software, and you are welcome to redistribute it
under certain conditions (GNU General Public License,
see ~abinit/COPYING or http://www.gnu.org/copyleft/gpl.txt).
ABINIT is a project of the Universite Catholique de Louvain,
Corning Inc. and other collaborators, see ~abinit/doc/developers/contributors.txt .
Please read https://docs.abinit.org/theory/acknowledgments for suggested
acknowledgments of the ABINIT effort.
For more information, see https://www.abinit.org .
.Starting date : Tue 10 Nov 2020.
- ( at 22h21 )
- input file -> /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/TestBot_MPI1/tutorespfn_teph4zpr_1-teph4zpr_2-teph4zpr_3-teph4zpr_4-teph4zpr_5-teph4zpr_6-teph4zpr_7-teph4zpr_8/teph4zpr_7.in
- output file -> teph4zpr_7.out
- root for input files -> teph4zpr_7i
- root for output files -> teph4zpr_7o
DATASET 1 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 1.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 20 mffmem = 1 mkmem = 8
mpw = 1304 nfft = 5832 nkpt = 8
================================================================================
P This job should need less than 6.411 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 3.186 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
DATASET 2 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 2.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 20 mffmem = 1 mkmem = 29
mpw = 1304 nfft = 5832 nkpt = 29
================================================================================
P This job should need less than 15.146 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 11.543 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
DATASET 3 : space group Fm -3 m (#225); Bravais cF (face-center cubic)
================================================================================
Values of the parameters that define the memory need for DATASET 3.
intxc = 0 ionmov = 0 iscf = 7 lmnmax = 5
lnmax = 5 mgfft = 18 mpssoang = 3 mqgrid = 3001
natom = 2 nloc_mem = 1 nspden = 1 nspinor = 1
nsppol = 1 nsym = 48 n1xccc = 2501 ntypat = 2
occopt = 1 xclevel = 2
- mband = 20 mffmem = 1 mkmem = 72
mpw = 1304 nfft = 5832 nkpt = 72
================================================================================
P This job should need less than 33.030 Mbytes of memory.
Rough estimation (10% accuracy) of disk space for files :
_ WF disk file : 28.654 Mbytes ; DEN or POT disk file : 0.046 Mbytes.
================================================================================
--------------------------------------------------------------------------------
------------- Echo of variables that govern the present computation ------------
--------------------------------------------------------------------------------
-
- outvars: echo of selected default values
- iomode0 = 0 , fftalg0 =312 , wfoptalg0 = 0
-
- outvars: echo of global parameters not present in the input file
- max_nthreads = 0
-
-outvars: echo values of preprocessed input variables --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
boxcutmin 1.10000000E+00
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_ngqpt_fine1 4 4 4
eph_ngqpt_fine2 8 8 8
eph_ngqpt_fine3 12 12 12
eph_stern 1
eph_task 4
- fftalg 312
istwfk1 2 0 3 0 0 0 7 0
istwfk2 2 0 0 0 3 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0
7 0 0 0 0 0 0 0 0
istwfk3 2 0 0 0 0 0 3 0 0 0
0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0
0 0 7 0 0 0 0 0 0 0
outvar_i_n : Printing only first 50 k-points.
ixc 11
jdtset 1 2 3
kpt1 0.00000000E+00 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
-2.50000000E-01 5.00000000E-01 2.50000000E-01
kpt2 0.00000000E+00 0.00000000E+00 0.00000000E+00
1.25000000E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
3.75000000E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
1.25000000E-01 1.25000000E-01 0.00000000E+00
2.50000000E-01 1.25000000E-01 0.00000000E+00
3.75000000E-01 1.25000000E-01 0.00000000E+00
5.00000000E-01 1.25000000E-01 0.00000000E+00
-3.75000000E-01 1.25000000E-01 0.00000000E+00
-2.50000000E-01 1.25000000E-01 0.00000000E+00
-1.25000000E-01 1.25000000E-01 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
3.75000000E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-3.75000000E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
3.75000000E-01 3.75000000E-01 0.00000000E+00
5.00000000E-01 3.75000000E-01 0.00000000E+00
-3.75000000E-01 3.75000000E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
3.75000000E-01 2.50000000E-01 1.25000000E-01
5.00000000E-01 2.50000000E-01 1.25000000E-01
-3.75000000E-01 2.50000000E-01 1.25000000E-01
5.00000000E-01 3.75000000E-01 1.25000000E-01
-3.75000000E-01 3.75000000E-01 1.25000000E-01
-2.50000000E-01 3.75000000E-01 1.25000000E-01
-3.75000000E-01 5.00000000E-01 1.25000000E-01
-2.50000000E-01 5.00000000E-01 2.50000000E-01
kpt3 0.00000000E+00 0.00000000E+00 0.00000000E+00
8.33333333E-02 0.00000000E+00 0.00000000E+00
1.66666667E-01 0.00000000E+00 0.00000000E+00
2.50000000E-01 0.00000000E+00 0.00000000E+00
3.33333333E-01 0.00000000E+00 0.00000000E+00
4.16666667E-01 0.00000000E+00 0.00000000E+00
5.00000000E-01 0.00000000E+00 0.00000000E+00
8.33333333E-02 8.33333333E-02 0.00000000E+00
1.66666667E-01 8.33333333E-02 0.00000000E+00
2.50000000E-01 8.33333333E-02 0.00000000E+00
3.33333333E-01 8.33333333E-02 0.00000000E+00
4.16666667E-01 8.33333333E-02 0.00000000E+00
5.00000000E-01 8.33333333E-02 0.00000000E+00
-4.16666667E-01 8.33333333E-02 0.00000000E+00
-3.33333333E-01 8.33333333E-02 0.00000000E+00
-2.50000000E-01 8.33333333E-02 0.00000000E+00
-1.66666667E-01 8.33333333E-02 0.00000000E+00
-8.33333333E-02 8.33333333E-02 0.00000000E+00
1.66666667E-01 1.66666667E-01 0.00000000E+00
2.50000000E-01 1.66666667E-01 0.00000000E+00
3.33333333E-01 1.66666667E-01 0.00000000E+00
4.16666667E-01 1.66666667E-01 0.00000000E+00
5.00000000E-01 1.66666667E-01 0.00000000E+00
-4.16666667E-01 1.66666667E-01 0.00000000E+00
-3.33333333E-01 1.66666667E-01 0.00000000E+00
-2.50000000E-01 1.66666667E-01 0.00000000E+00
-1.66666667E-01 1.66666667E-01 0.00000000E+00
2.50000000E-01 2.50000000E-01 0.00000000E+00
3.33333333E-01 2.50000000E-01 0.00000000E+00
4.16666667E-01 2.50000000E-01 0.00000000E+00
5.00000000E-01 2.50000000E-01 0.00000000E+00
-4.16666667E-01 2.50000000E-01 0.00000000E+00
-3.33333333E-01 2.50000000E-01 0.00000000E+00
-2.50000000E-01 2.50000000E-01 0.00000000E+00
3.33333333E-01 3.33333333E-01 0.00000000E+00
4.16666667E-01 3.33333333E-01 0.00000000E+00
5.00000000E-01 3.33333333E-01 0.00000000E+00
-4.16666667E-01 3.33333333E-01 0.00000000E+00
-3.33333333E-01 3.33333333E-01 0.00000000E+00
4.16666667E-01 4.16666667E-01 0.00000000E+00
5.00000000E-01 4.16666667E-01 0.00000000E+00
-4.16666667E-01 4.16666667E-01 0.00000000E+00
5.00000000E-01 5.00000000E-01 0.00000000E+00
2.50000000E-01 1.66666667E-01 8.33333333E-02
3.33333333E-01 1.66666667E-01 8.33333333E-02
4.16666667E-01 1.66666667E-01 8.33333333E-02
5.00000000E-01 1.66666667E-01 8.33333333E-02
-4.16666667E-01 1.66666667E-01 8.33333333E-02
3.33333333E-01 2.50000000E-01 8.33333333E-02
4.16666667E-01 2.50000000E-01 8.33333333E-02
outvar_i_n : Printing only first 50 k-points.
kptrlatt1 4 0 0 0 4 0 0 0 4
kptrlatt2 8 0 0 0 8 0 0 0 8
kptrlatt3 12 0 0 0 12 0 0 0 12
kptrlen1 2.27305763E+01
kptrlen2 4.54611525E+01
kptrlen3 6.81917288E+01
mixprec 1
P mkmem1 8
P mkmem2 29
P mkmem3 72
natom 2
nband1 20
nband2 20
nband3 20
ndtset 3
ngfft 18 18 18
nkpt1 8
nkpt2 29
nkpt3 72
nkptgw 1
nline 100
nsym 48
ntypat 2
occ1 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
optdriver 7
prtphdos 0
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tolwfr 1.00000000E-16
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
wtk1 0.01563 0.12500 0.06250 0.09375 0.37500 0.18750
0.04688 0.09375
wtk2 0.00195 0.01563 0.01563 0.01563 0.00781 0.01172
0.04688 0.04688 0.04688 0.04688 0.04688 0.02344
0.01172 0.04688 0.04688 0.04688 0.02344 0.01172
0.04688 0.02344 0.00586 0.04688 0.09375 0.04688
0.04688 0.09375 0.04688 0.02344 0.01172
wtk3 0.00058 0.00463 0.00463 0.00463 0.00463 0.00463
0.00231 0.00347 0.01389 0.01389 0.01389 0.01389
0.01389 0.01389 0.01389 0.01389 0.01389 0.00694
0.00347 0.01389 0.01389 0.01389 0.01389 0.01389
0.01389 0.01389 0.00694 0.00347 0.01389 0.01389
0.01389 0.01389 0.01389 0.00694 0.00347 0.01389
0.01389 0.01389 0.00694 0.00347 0.01389 0.00694
0.00174 0.01389 0.02778 0.02778 0.02778 0.01389
0.01389 0.02778
outvars : Printing only first 50 k-points.
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
chkinp: Checking input parameters for consistency, jdtset= 1.
chkinp: Checking input parameters for consistency, jdtset= 2.
chkinp: Checking input parameters for consistency, jdtset= 3.
================================================================================
== DATASET 1 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 1, }
dimensions: {natom: 2, nkpt: 8, mband: 20, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS1_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS1_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--- Pseudopotential description ------------------------------------------------
- pspini: atom type 1 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/O.psp8
- O ONCVPSP r_core= 1.36 1.46 1.26
- 8.00000 6.00000 151103 znucl, zion, pspdat
8 11 2 4 600 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
5.99000000000000 4.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2 1
extension_switch 1
pspatm : epsatm= 6.19401560
--- l ekb(1:nproj) -->
0 5.257212 0.704241
1 -5.135443 -1.451781
2 -4.371486
pspatm: atomic psp has been read and splines computed
- pspini: atom type 2 psp file is /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- pspatm: opening atomic psp file /home/buildbot/ABINIT/alps_gnu_9.3_openmpi/trunk_rel2dev/tests/Psps_for_tests/Mg-sp-gw.psp8
- Mg ONCVPSP r_core= 1.16 1.56
- 12.00000 10.00000 150902 znucl, zion, pspdat
8 11 1 4 400 0.00000 pspcod,pspxc,lmax,lloc,mmax,r2well
3.99000000000000 0.00000000000000 0.00000000000000 rchrg,fchrg,qchrg
nproj 2 2
extension_switch 1
pspatm : epsatm= 9.22508734
--- l ekb(1:nproj) -->
0 2.568049 -0.805950
1 -6.605901 -2.664074
pspatm: atomic psp has been read and splines computed
2.46705647E+02 ecore*ucvol(ha*bohr**3)
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 7.721 3.231 1.248
100.0 0.009 7.656 3.167 1.312
200.0 0.017 7.592 3.102 1.377
300.0 0.026 7.527 3.037 1.442
Number of bands in e-ph self-energy sum: 20
From bsum_start: 1 to bsum_stop: 20
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [4, 4, 4]
Number of q-points in the IBZ: 8
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 20
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 8 of 8
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
DVDB file contains all q-points in the IBZ --> Reading DFPT potentials from file.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 7.721
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.570 0.080 0.097 -0.002 0.823 -3.999 4.096 0.000 0.000
7 4.490 4.570 0.080 0.097 -0.002 0.823 -3.999 4.096 0.000 0.000
8 4.490 4.570 0.080 0.097 -0.002 0.823 -3.999 4.096 0.000 0.000
9 8.969 8.888 -0.081 -0.083 -0.000 0.981 -0.055 -0.028 4.479 4.318
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.318 (OTMS: 4.299)
QP_gap - KS_gap: -0.161 (OTMS: -0.180)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 7.656
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.570 0.080 0.097 -0.002 0.822 -4.089 4.187 0.000 0.000
7 4.490 4.570 0.080 0.097 -0.002 0.822 -4.089 4.187 0.000 0.000
8 4.490 4.570 0.080 0.097 -0.002 0.822 -4.089 4.187 0.000 0.000
9 8.969 8.887 -0.082 -0.083 -0.000 0.981 -0.051 -0.033 4.479 4.317
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.317 (OTMS: 4.298)
QP_gap - KS_gap: -0.162 (OTMS: -0.181)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 7.592
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.572 0.082 0.102 -0.002 0.809 -4.736 4.837 0.000 0.000
7 4.490 4.572 0.082 0.102 -0.002 0.809 -4.736 4.837 0.000 0.000
8 4.490 4.572 0.082 0.102 -0.002 0.809 -4.736 4.837 0.000 0.000
9 8.969 8.879 -0.090 -0.092 -0.000 0.979 -0.036 -0.055 4.479 4.307
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.307 (OTMS: 4.286)
QP_gap - KS_gap: -0.172 (OTMS: -0.193)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 7.527
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.579 0.089 0.114 -0.003 0.783 -5.813 5.927 0.000 0.000
7 4.490 4.579 0.089 0.114 -0.003 0.783 -5.813 5.927 0.000 0.000
8 4.490 4.579 0.089 0.114 -0.003 0.783 -5.813 5.927 0.000 0.000
9 8.969 8.864 -0.105 -0.108 -0.000 0.974 -0.024 -0.083 4.479 4.285
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.285 (OTMS: 4.258)
QP_gap - KS_gap: -0.194 (OTMS: -0.221)
============================================================================================
================================================================================
== DATASET 2 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 2, }
dimensions: {natom: 2, nkpt: 29, mband: 20, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS2_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS2_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 5.900 1.410 3.069
100.0 0.009 5.948 1.459 3.020
200.0 0.017 5.997 1.507 2.972
300.0 0.026 6.045 1.556 2.923
Number of bands in e-ph self-energy sum: 20
From bsum_start: 1 to bsum_stop: 20
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [8, 8, 8]
Number of q-points in the IBZ: 29
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 20
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 29 of 29
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
Cannot find eph_ngqpt_fine q-points in DVDB --> Activating Fourier interpolation.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 5.900
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.594 0.104 0.159 -0.005 0.654 -4.024 4.183 0.000 0.000
7 4.490 4.594 0.104 0.159 -0.005 0.654 -4.024 4.183 0.000 0.000
8 4.490 4.594 0.104 0.159 -0.005 0.654 -4.024 4.183 0.000 0.000
9 8.969 8.872 -0.097 -0.100 -0.000 0.965 -0.071 -0.029 4.479 4.278
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.278 (OTMS: 4.220)
QP_gap - KS_gap: -0.201 (OTMS: -0.259)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 5.948
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.594 0.104 0.160 -0.005 0.651 -4.177 4.337 0.000 0.000
7 4.490 4.594 0.104 0.160 -0.005 0.651 -4.177 4.337 0.000 0.000
8 4.490 4.594 0.104 0.160 -0.005 0.651 -4.177 4.337 0.000 0.000
9 8.969 8.871 -0.098 -0.101 -0.000 0.964 -0.066 -0.035 4.479 4.277
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.277 (OTMS: 4.218)
QP_gap - KS_gap: -0.202 (OTMS: -0.261)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 5.997
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.593 0.103 0.170 -0.008 0.607 -4.941 5.110 0.000 0.000
7 4.490 4.593 0.103 0.170 -0.008 0.607 -4.941 5.110 0.000 0.000
8 4.490 4.593 0.103 0.170 -0.008 0.607 -4.941 5.110 0.000 0.000
9 8.969 8.863 -0.106 -0.110 -0.000 0.961 -0.050 -0.060 4.479 4.270
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.270 (OTMS: 4.199)
QP_gap - KS_gap: -0.209 (OTMS: -0.280)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 6.045
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.588 0.099 0.200 -0.020 0.492 -6.128 6.329 0.000 0.000
7 4.490 4.588 0.099 0.200 -0.020 0.492 -6.128 6.329 0.000 0.000
8 4.490 4.588 0.099 0.200 -0.020 0.492 -6.128 6.329 0.000 0.000
9 8.969 8.846 -0.122 -0.128 -0.000 0.955 -0.038 -0.090 4.479 4.258
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.258 (OTMS: 4.151)
QP_gap - KS_gap: -0.221 (OTMS: -0.328)
============================================================================================
================================================================================
== DATASET 3 ==================================================================
- mpi_nproc: 1, omp_nthreads: -1 (-1 if OMP is not activated)
--- !DatasetInfo
iteration_state: {dtset: 3, }
dimensions: {natom: 2, nkpt: 72, mband: 20, nsppol: 1, nspinor: 1, nspden: 1, mpw: 1304, }
cutoff_energies: {ecut: 35.0, pawecutdg: -1.0, }
electrons: {nelect: 1.60000000E+01, charge: 0.00000000E+00, occopt: 1.00000000E+00, tsmear: 1.00000000E-02, }
meta: {optdriver: 7, eph_task: 4, }
...
mkfilename: getwfk from: teph4zpr_3o_DS3_WFK
mkfilename: getddb from: teph4zpr_1_DDB
mkfilename: getpot from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
mkfilename: getdvdb from: teph4zpr_2_DVDB
Exchange-correlation functional for the present dataset will be:
GGA: Perdew-Burke-Ernzerhof functional - ixc=11
Citation for XC functional:
J.P.Perdew, K.Burke, M.Ernzerhof, PRL 77, 3865 (1996)
- Reading GS states from WFK file: teph4zpr_3o_DS3_WFK
- Reading DDB from file: teph4zpr_1_DDB
- Reading DVDB from file: teph4zpr_2_DVDB
Real(R)+Recip(G) space primitive vectors, cartesian coordinates (Bohr,Bohr^-1):
R(1)= 0.0000000 4.0182362 4.0182362 G(1)= -0.1244327 0.1244327 0.1244327
R(2)= 4.0182362 0.0000000 4.0182362 G(2)= 0.1244327 -0.1244327 0.1244327
R(3)= 4.0182362 4.0182362 0.0000000 G(3)= 0.1244327 0.1244327 -0.1244327
Unit cell volume ucvol= 1.2975866E+02 bohr^3
Angles (23,13,12)= 6.00000000E+01 6.00000000E+01 6.00000000E+01 degrees
Now the whole DDB is in central memory
================================================================================
Dielectric Tensor and Effective Charges
anaddb : Zero the imaginary part of the Dynamical Matrix at Gamma,
and impose the ASR on the effective charges
The violation of the charge neutrality conditions
by the effective charges is as follows :
atom electric field
displacement direction
1 1 -0.085945 0.000000
1 2 0.000000 0.000000
1 3 0.000000 0.000000
2 1 0.000000 0.000000
2 2 -0.085945 0.000000
2 3 -0.000000 0.000000
3 1 -0.000000 0.000000
3 2 -0.000000 0.000000
3 3 -0.085945 0.000000
Effective charge tensors after
imposition of the charge neutrality,
and eventual restriction to some part :
atom displacement
1 1 2.040887E+00 -1.659033E-16 -1.658285E-16
1 2 -1.659033E-16 2.040887E+00 1.659781E-16
1 3 1.659033E-16 1.659033E-16 2.040887E+00
2 1 -2.040887E+00 1.659033E-16 1.658285E-16
2 2 1.659033E-16 -2.040887E+00 -1.659781E-16
2 3 -1.659033E-16 -1.659033E-16 -2.040887E+00
Now, the imaginary part of the dynamical matrix is zeroed
- Found dielectric tensor and Born effective charges in DDB file: teph4zpr_1_DDB
- Cannot find quadrupole tensor in DDB file: teph4zpr_1_DDB
Values initialized with zeros.
Homogeneous q point set in the B.Z.
Grid q points : 64
1) 0.00000000E+00 0.00000000E+00 0.00000000E+00
2) 2.50000000E-01 0.00000000E+00 0.00000000E+00
3) 5.00000000E-01 0.00000000E+00 0.00000000E+00
4) -2.50000000E-01 0.00000000E+00 0.00000000E+00
5) 0.00000000E+00 2.50000000E-01 0.00000000E+00
6) 2.50000000E-01 2.50000000E-01 0.00000000E+00
7) 5.00000000E-01 2.50000000E-01 0.00000000E+00
8) -2.50000000E-01 2.50000000E-01 0.00000000E+00
9) 0.00000000E+00 5.00000000E-01 0.00000000E+00
10) 2.50000000E-01 5.00000000E-01 0.00000000E+00
11) 5.00000000E-01 5.00000000E-01 0.00000000E+00
12) -2.50000000E-01 5.00000000E-01 0.00000000E+00
13) 0.00000000E+00 -2.50000000E-01 0.00000000E+00
14) 2.50000000E-01 -2.50000000E-01 0.00000000E+00
15) 5.00000000E-01 -2.50000000E-01 0.00000000E+00
16) -2.50000000E-01 -2.50000000E-01 0.00000000E+00
17) 0.00000000E+00 0.00000000E+00 2.50000000E-01
18) 2.50000000E-01 0.00000000E+00 2.50000000E-01
19) 5.00000000E-01 0.00000000E+00 2.50000000E-01
20) -2.50000000E-01 0.00000000E+00 2.50000000E-01
21) 0.00000000E+00 2.50000000E-01 2.50000000E-01
22) 2.50000000E-01 2.50000000E-01 2.50000000E-01
23) 5.00000000E-01 2.50000000E-01 2.50000000E-01
24) -2.50000000E-01 2.50000000E-01 2.50000000E-01
25) 0.00000000E+00 5.00000000E-01 2.50000000E-01
26) 2.50000000E-01 5.00000000E-01 2.50000000E-01
27) 5.00000000E-01 5.00000000E-01 2.50000000E-01
28) -2.50000000E-01 5.00000000E-01 2.50000000E-01
29) 0.00000000E+00 -2.50000000E-01 2.50000000E-01
30) 2.50000000E-01 -2.50000000E-01 2.50000000E-01
31) 5.00000000E-01 -2.50000000E-01 2.50000000E-01
32) -2.50000000E-01 -2.50000000E-01 2.50000000E-01
33) 0.00000000E+00 0.00000000E+00 5.00000000E-01
34) 2.50000000E-01 0.00000000E+00 5.00000000E-01
35) 5.00000000E-01 0.00000000E+00 5.00000000E-01
36) -2.50000000E-01 0.00000000E+00 5.00000000E-01
37) 0.00000000E+00 2.50000000E-01 5.00000000E-01
38) 2.50000000E-01 2.50000000E-01 5.00000000E-01
39) 5.00000000E-01 2.50000000E-01 5.00000000E-01
40) -2.50000000E-01 2.50000000E-01 5.00000000E-01
41) 0.00000000E+00 5.00000000E-01 5.00000000E-01
42) 2.50000000E-01 5.00000000E-01 5.00000000E-01
43) 5.00000000E-01 5.00000000E-01 5.00000000E-01
44) -2.50000000E-01 5.00000000E-01 5.00000000E-01
45) 0.00000000E+00 -2.50000000E-01 5.00000000E-01
46) 2.50000000E-01 -2.50000000E-01 5.00000000E-01
47) 5.00000000E-01 -2.50000000E-01 5.00000000E-01
48) -2.50000000E-01 -2.50000000E-01 5.00000000E-01
49) 0.00000000E+00 0.00000000E+00 -2.50000000E-01
50) 2.50000000E-01 0.00000000E+00 -2.50000000E-01
51) 5.00000000E-01 0.00000000E+00 -2.50000000E-01
52) -2.50000000E-01 0.00000000E+00 -2.50000000E-01
53) 0.00000000E+00 2.50000000E-01 -2.50000000E-01
54) 2.50000000E-01 2.50000000E-01 -2.50000000E-01
55) 5.00000000E-01 2.50000000E-01 -2.50000000E-01
56) -2.50000000E-01 2.50000000E-01 -2.50000000E-01
57) 0.00000000E+00 5.00000000E-01 -2.50000000E-01
58) 2.50000000E-01 5.00000000E-01 -2.50000000E-01
59) 5.00000000E-01 5.00000000E-01 -2.50000000E-01
60) -2.50000000E-01 5.00000000E-01 -2.50000000E-01
61) 0.00000000E+00 -2.50000000E-01 -2.50000000E-01
62) 2.50000000E-01 -2.50000000E-01 -2.50000000E-01
63) 5.00000000E-01 -2.50000000E-01 -2.50000000E-01
64) -2.50000000E-01 -2.50000000E-01 -2.50000000E-01
The interatomic forces have been obtained
--------------------------------------------------------------------------------
=== Gaps, band edges and relative position wrt Fermi level ===
Direct band gap semiconductor
Fundamental gap: 4.479 (eV)
VBM: 4.490 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
CBM: 8.969 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Direct gap: 4.479 (eV) at k: [ 0.0000E+00, 0.0000E+00, 0.0000E+00]
Position of CBM/VBM with respect to the Fermi level:
Notations: mu_e = Fermi level, D_v = (mu_e - VBM), D_c = (CBM - mu_e)
T(K) kT (eV) mu_e (eV) D_v (eV) D_c (eV)
0.0 0.000 5.900 1.410 3.069
100.0 0.009 5.948 1.459 3.020
200.0 0.017 5.997 1.507 2.972
300.0 0.026 6.045 1.556 2.923
Number of bands in e-ph self-energy sum: 20
From bsum_start: 1 to bsum_stop: 20
Treating high-energy bands with Sternheimer and static self-energy.
Tolwfr: 1.000000E-16, nline: 100
Symsigma: 1 Timrev: 1
Imaginary shift in the denominator (zcut): 0.010 [eV]
Method for q-space integration: Standard quadrature
Both Real and Imaginary part of Sigma will be computed.
Number of frequencies along the real axis: 0 , Step: 0.000 [eV]
Number of frequency in generalized Eliashberg functions: 0
Number of temperatures: 4 From: 0.000000E+00 to 3.000000E+02 [K]
Ab-initio q-mesh from DDB file: [4, 4, 4]
Q-mesh used for self-energy integration [ngqpt]: [12, 12, 12]
Number of q-points in the IBZ: 72
asr: 1 chneut: 1
dipdip: 1 symdynmat: 1
Number of k-points for self-energy corrections: 1
Including all final {mk+q} states inside energy window: [4.347 9.112 ] [eV]
List of k-points for self-energy corrections:
1 1 [ 0.0000E+00, 0.0000E+00, 0.0000E+00] 6 9
=== MPI parallelism ===
P Allocating and summing bands from my_bsum_start: 1 up to my_bsum_stop: 20
P Number of CPUs for parallelism over perturbations: 1
P Number of perturbations treated by this CPU: 6
P Number of CPUs for parallelism over q-points: 1
P Number of q-points in the IBZ treated by this proc: 72 of 72
P Number of CPUs for parallelism over bands: 1
P Number of CPUs for parallelism over spins: 1
P Number of CPUs for parallelism over k-points: 1
P Number of k-point in Sigma_nk treated by this proc: 1 of 1
Reading GS KS potential for Sternheimer from: MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc
Cannot find eph_ngqpt_fine q-points in DVDB --> Activating Fourier interpolation.
================================================================================
Final results in eV.
Notations:
eKS: Kohn-Sham energy. eQP: quasi-particle energy.
eQP - eKS: Difference between the QP and the KS energy.
SE1(eKS): Real part of the self-energy computed at the KS energy, SE2 for imaginary part.
Z(eKS): Renormalization factor.
FAN: Real part of the Fan term at eKS. DW: Debye-Waller term.
DeKS: KS energy difference between this band and band-1, DeQP same meaning but for eQP.
OTMS: On-the-mass-shell approximation with eQP ~= eKS + Sigma(omega=eKS)
TAU(eKS): Lifetime in femtoseconds computed at the KS energy.
mu_e: Fermi level for given (T, nelect)
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 0.0 [K], mu_e: 5.900
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.598 0.108 0.194 -0.008 0.557 -4.007 4.201 0.000 0.000
7 4.490 4.598 0.108 0.194 -0.008 0.557 -4.007 4.201 0.000 0.000
8 4.490 4.598 0.108 0.194 -0.008 0.557 -4.007 4.201 0.000 0.000
9 8.969 8.862 -0.107 -0.114 -0.001 0.939 -0.084 -0.030 4.479 4.264
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.264 (OTMS: 4.171)
QP_gap - KS_gap: -0.215 (OTMS: -0.308)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 100.0 [K], mu_e: 5.948
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.598 0.108 0.195 -0.008 0.552 -4.192 4.387 0.000 0.000
7 4.490 4.598 0.108 0.195 -0.008 0.552 -4.192 4.387 0.000 0.000
8 4.490 4.598 0.108 0.195 -0.008 0.552 -4.192 4.387 0.000 0.000
9 8.969 8.861 -0.108 -0.115 -0.001 0.938 -0.079 -0.036 4.479 4.263
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.263 (OTMS: 4.169)
QP_gap - KS_gap: -0.216 (OTMS: -0.310)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 200.0 [K], mu_e: 5.997
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.597 0.107 0.202 -0.010 0.530 -5.007 5.208 0.000 0.000
7 4.490 4.597 0.107 0.202 -0.010 0.530 -5.007 5.208 0.000 0.000
8 4.490 4.597 0.107 0.202 -0.010 0.530 -5.007 5.208 0.000 0.000
9 8.969 8.853 -0.116 -0.124 -0.001 0.934 -0.063 -0.062 4.479 4.256
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.256 (OTMS: 4.153)
QP_gap - KS_gap: -0.223 (OTMS: -0.326)
============================================================================================
K-point: [ 0.0000E+00, 0.0000E+00, 0.0000E+00], T: 300.0 [K], mu_e: 6.045
B eKS eQP eQP-eKS SE1(eKS) SE2(eKS) Z(eKS) FAN(eKS) DW DeKS DeQP
6 4.490 4.594 0.104 0.218 -0.019 0.479 -6.257 6.475 0.000 0.000
7 4.490 4.594 0.104 0.218 -0.019 0.479 -6.257 6.475 0.000 0.000
8 4.490 4.594 0.104 0.218 -0.019 0.479 -6.257 6.475 0.000 0.000
9 8.969 8.836 -0.133 -0.143 -0.001 0.925 -0.051 -0.092 4.479 4.242
KS gap: 4.479 (assuming bval:8 ==> bcond:9)
QP gap: 4.242 (OTMS: 4.118)
QP_gap - KS_gap: -0.237 (OTMS: -0.361)
============================================================================================
== END DATASET(S) ==============================================================
================================================================================
-outvars: echo values of variables after computation --------
acell 1.0000000000E+00 1.0000000000E+00 1.0000000000E+00 Bohr
amu 1.59994000E+01 2.43050000E+01
bdgw 8 9
boxcutmin 1.10000000E+00
ddb_ngqpt 4 4 4
ecut 3.50000000E+01 Hartree
eph_intmeth 1
eph_ngqpt_fine1 4 4 4
eph_ngqpt_fine2 8 8 8
eph_ngqpt_fine3 12 12 12
eph_stern 1
eph_task 4
etotal1 0.0000000000E+00
etotal2 0.0000000000E+00
etotal3 0.0000000000E+00
fcart1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
fcart3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
- fftalg 312
ixc 11
jdtset 1 2 3
kptrlatt1 4 0 0 0 4 0 0 0 4
kptrlatt2 8 0 0 0 8 0 0 0 8
kptrlatt3 12 0 0 0 12 0 0 0 12
kptrlen1 2.27305763E+01
kptrlen2 4.54611525E+01
kptrlen3 6.81917288E+01
mixprec 1
P mkmem1 8
P mkmem2 29
P mkmem3 72
natom 2
nband1 20
nband2 20
nband3 20
ndtset 3
ngfft 18 18 18
nkpt1 8
nkpt2 29
nkpt3 72
nkptgw 1
nline 100
nsym 48
ntypat 2
occ1 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ2 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
occ3 2.000000 2.000000 2.000000 2.000000 2.000000 2.000000
2.000000 2.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000
optdriver 7
prtphdos 0
rprim 0.0000000000E+00 4.0182361526E+00 4.0182361526E+00
4.0182361526E+00 0.0000000000E+00 4.0182361526E+00
4.0182361526E+00 4.0182361526E+00 0.0000000000E+00
spgroup 225
strten1 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten2 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
strten3 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
symrel 1 0 0 0 1 0 0 0 1 -1 0 0 0 -1 0 0 0 -1
0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0
-1 0 0 -1 0 1 -1 1 0 1 0 0 1 0 -1 1 -1 0
0 1 -1 1 0 -1 0 0 -1 0 -1 1 -1 0 1 0 0 1
-1 0 0 -1 1 0 -1 0 1 1 0 0 1 -1 0 1 0 -1
0 -1 1 1 -1 0 0 -1 0 0 1 -1 -1 1 0 0 1 0
1 0 0 0 0 1 0 1 0 -1 0 0 0 0 -1 0 -1 0
0 1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1
-1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1 0 0
0 -1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1
1 0 -1 0 0 -1 0 1 -1 -1 0 1 0 0 1 0 -1 1
0 1 0 0 0 1 1 0 0 0 -1 0 0 0 -1 -1 0 0
1 0 -1 0 1 -1 0 0 -1 -1 0 1 0 -1 1 0 0 1
0 -1 0 0 -1 1 1 -1 0 0 1 0 0 1 -1 -1 1 0
-1 0 1 -1 0 0 -1 1 0 1 0 -1 1 0 0 1 -1 0
0 1 0 1 0 0 0 0 1 0 -1 0 -1 0 0 0 0 -1
0 0 -1 0 1 -1 1 0 -1 0 0 1 0 -1 1 -1 0 1
1 -1 0 0 -1 1 0 -1 0 -1 1 0 0 1 -1 0 1 0
0 0 1 1 0 0 0 1 0 0 0 -1 -1 0 0 0 -1 0
-1 1 0 -1 0 0 -1 0 1 1 -1 0 1 0 0 1 0 -1
0 0 1 0 1 0 1 0 0 0 0 -1 0 -1 0 -1 0 0
1 -1 0 0 -1 0 0 -1 1 -1 1 0 0 1 0 0 1 -1
0 0 -1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1
-1 1 0 -1 0 1 -1 0 0 1 -1 0 1 0 -1 1 0 0
tolwfr 1.00000000E-16
tmesh 0.00000000E+00 1.00000000E+02 4.00000000E+00
typat 2 1
xangst 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
2.1263589907E+00 2.1263589907E+00 2.1263589907E+00
xcart 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
4.0182361526E+00 4.0182361526E+00 4.0182361526E+00
xred 0.0000000000E+00 0.0000000000E+00 0.0000000000E+00
5.0000000000E-01 5.0000000000E-01 5.0000000000E-01
zcut 3.67493254E-04 Hartree
znucl 8.00000 12.00000
================================================================================
- Timing analysis has been suppressed with timopt=0
================================================================================
Suggested references for the acknowledgment of ABINIT usage.
The users of ABINIT have little formal obligations with respect to the ABINIT group
(those specified in the GNU General Public License, http://www.gnu.org/copyleft/gpl.txt).
However, it is common practice in the scientific literature,
to acknowledge the efforts of people that have made the research possible.
In this spirit, please find below suggested citations of work written by ABINIT developers,
corresponding to implementations inside of ABINIT that you have used in the present run.
Note also that it will be of great value to readers of publications presenting these results,
to read papers enabling them to understand the theoretical formalism and details
of the ABINIT implementation.
For information on why they are suggested, see also https://docs.abinit.org/theory/acknowledgments.
-
- [1] The Abinit project: Impact, environment and recent developments.
- Computer Phys. Comm. 248, 107042 (2020).
- X.Gonze, B. Amadon, G. Antonius, F.Arnardi, L.Baguet, J.-M.Beuken,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, N.Brouwer, F.Bruneval,
- G.Brunin, T.Cavignac, J.-B. Charraud, Wei Chen, M.Cote, S.Cottenier,
- J.Denier, G.Geneste, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, Xu He, N.Helbig, N.Holzwarth, Y.Jia, F.Jollet,
- W.Lafargue-Dit-Hauret, K.Lejaeghere, M.A.L.Marques, A.Martin, C.Martins,
- H.P.C. Miranda, F.Naccarato, K. Persson, G.Petretto, V.Planes, Y.Pouillon,
- S.Prokhorenko, F.Ricci, G.-M.Rignanese, A.H.Romero, M.M.Schmitt, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, G.Zerah and J.W.Zwanzig
- Comment: the fifth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT20.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2020
-
- [2] Optimized norm-conserving Vanderbilt pseudopotentials.
- D.R. Hamann, Phys. Rev. B 88, 085117 (2013).
- Comment: Some pseudopotential generated using the ONCVPSP code were used.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#hamann2013
-
- [3] ABINIT: Overview, and focus on selected capabilities
- J. Chem. Phys. 152, 124102 (2020).
- A. Romero, D.C. Allan, B. Amadon, G. Antonius, T. Applencourt, L.Baguet,
- J.Bieder, F.Bottin, J.Bouchet, E.Bousquet, F.Bruneval,
- G.Brunin, D.Caliste, M.Cote,
- J.Denier, C. Dreyer, Ph.Ghosez, M.Giantomassi, Y.Gillet, O.Gingras,
- D.R.Hamann, G.Hautier, F.Jollet, G. Jomard,
- A.Martin,
- H.P.C. Miranda, F.Naccarato, G.Petretto, N.A. Pike, V.Planes,
- S.Prokhorenko, T. Rangel, F.Ricci, G.-M.Rignanese, M.Royo, M.Stengel, M.Torrent,
- M.J.van Setten, B.Van Troeye, M.J.Verstraete, J.Wiktor, J.W.Zwanziger, and X.Gonze.
- Comment: a global overview of ABINIT, with focus on selected capabilities .
- Note that a version of this paper, that is not formatted for J. Chem. Phys
- is available at https://www.abinit.org/sites/default/files/ABINIT20_JPC.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#romero2020
-
- [4] Recent developments in the ABINIT software package.
- Computer Phys. Comm. 205, 106 (2016).
- X.Gonze, F.Jollet, F.Abreu Araujo, D.Adams, B.Amadon, T.Applencourt,
- C.Audouze, J.-M.Beuken, J.Bieder, A.Bokhanchuk, E.Bousquet, F.Bruneval
- D.Caliste, M.Cote, F.Dahm, F.Da Pieve, M.Delaveau, M.Di Gennaro,
- B.Dorado, C.Espejo, G.Geneste, L.Genovese, A.Gerossier, M.Giantomassi,
- Y.Gillet, D.R.Hamann, L.He, G.Jomard, J.Laflamme Janssen, S.Le Roux,
- A.Levitt, A.Lherbier, F.Liu, I.Lukacevic, A.Martin, C.Martins,
- M.J.T.Oliveira, S.Ponce, Y.Pouillon, T.Rangel, G.-M.Rignanese,
- A.H.Romero, B.Rousseau, O.Rubel, A.A.Shukri, M.Stankovski, M.Torrent,
- M.J.Van Setten, B.Van Troeye, M.J.Verstraete, D.Waroquier, J.Wiktor,
- B.Xu, A.Zhou, J.W.Zwanziger.
- Comment: the fourth generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT16.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2016
-
- And optionally:
-
- [5] ABINIT: First-principles approach of materials and nanosystem properties.
- Computer Phys. Comm. 180, 2582-2615 (2009).
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval,
- D. Caliste, R. Caracas, M. Cote, T. Deutsch, L. Genovese, Ph. Ghosez, M. Giantomassi
- S. Goedecker, D.R. Hamann, P. Hermet, F. Jollet, G. Jomard, S. Leroux, M. Mancini, S. Mazevet,
- M.J.T. Oliveira, G. Onida, Y. Pouillon, T. Rangel, G.-M. Rignanese, D. Sangalli, R. Shaltaf,
- M. Torrent, M.J. Verstraete, G. Zerah, J.W. Zwanziger
- Comment: the third generic paper describing the ABINIT project.
- Note that a version of this paper, that is not formatted for Computer Phys. Comm.
- is available at https://www.abinit.org/sites/default/files/ABINIT_CPC_v10.pdf .
- The licence allows the authors to put it on the Web.
- DOI and bibtex: see https://docs.abinit.org/theory/bibliography/#gonze2009
-
- Proc. 0 individual time (sec): cpu= 38.4 wall= 38.6
================================================================================
Calculation completed.
.Delivered 7 WARNINGs and 13 COMMENTs to log file.
+Overall time at end (sec) : cpu= 38.4 wall= 38.6
Now use:
abicomp.py sigeph teph4zpr_7o_DS*_SIGEPH.nc -e
to analyze the convergence with the \qq-mesh:
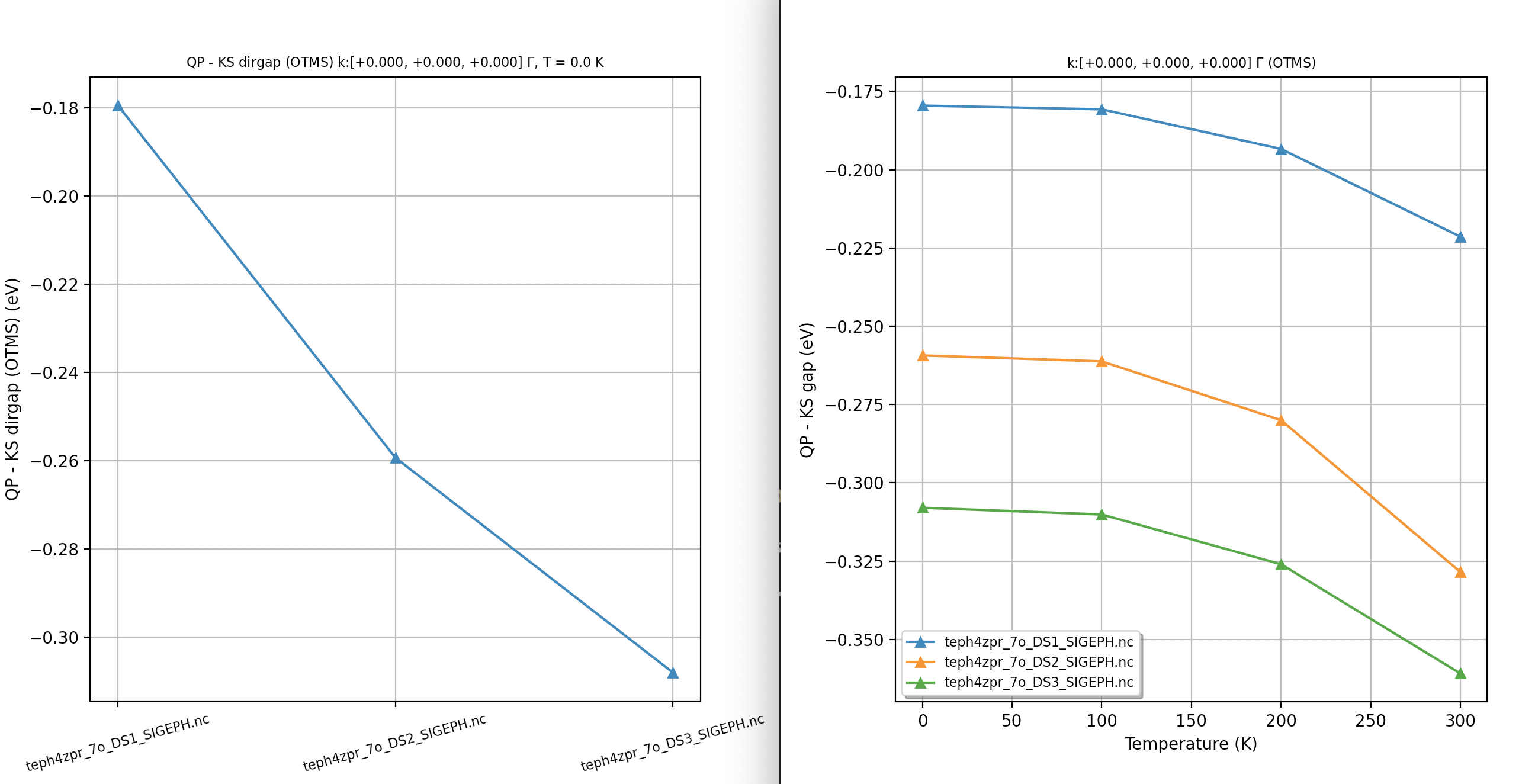
The convergence of the ZPR is shown in the left panel while the T-dependent band gaps obtained with different \qq-meshes are reported in the right panel.
Obviously we are still far from convergence but a 12x12x12 \qq-mesh is the best we can afford in a tutorial. It is clear that one should test denser \qq-meshes and, last but not least, monitor the behaviour for zcut → 0. These additional convergence tests cannot be covered in this lesson and they are left as exercises.
How to compute the spectral function¶
To compute the spectral function, we need to specify the number of frequencies via nfreqsp. The code will compute A_\nk(\ww) on a linear frequency mesh centered on the KS eigenvalue that spans the interval [\ee_\nk - \Delta, \ee_\nk + \Delta] with \Delta given by freqspmax. An odd number of frequency points is enforced by the code.
In a nutshell, one should add e.g.:
nfreqsp 301 freqspmax 1.0 eV
An example of input file is available here:
# Computation of spectral function with/without Sternheimer method ndtset 2 nfreqsp 201 freqspmax 2 eV prtvol 1 # This is for the automatic tests. Do not use in production! nband 20 eph_stern2 1 getpot_filepath2 "MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_POT.nc" ngkpt 12 12 12 eph_ngqpt_fine 12 12 12 getwfk_filepath "teph4zpr_3o_DS3_WFK" getddb_filepath "teph4zpr_1_DDB" ddb_ngqpt 4 4 4 getdvdb_filepath "teph4zpr_2_DVDB" ecut 35.0 nshiftk 1 shiftk 0 0 0 mixprec 1 boxcutmin 1.1 # Important to speedup the calculation and reduce memory. ############################################## #### SECTION: eph ############################################## optdriver 7 eph_task 4 tmesh 0 100 4 # [start, step, num_T] in K prtphdos 0 # Disable computation of PHDOS to save some time ############################################## #### SECTION: Sigma_nk ############################################## nkptgw 1 kptgw 0.0 0.0 0.0 bdgw 8 9 zcut 0.01 eV pp_dirpath = "$ABI_PSPDIR" pseudos = "O.psp8, Mg-sp-gw.psp8" structure = "abifile:MgO_eph_zpr/flow_zpr_mgo/w0/t0/outdata/out_DEN.nc" ############################################################## # This section is used only for regression testing of ABINIT # ############################################################## #%%<BEGIN TEST_INFO> #%% [setup] #%% executable = abinit #%% test_chain = teph4zpr_1.abi, teph4zpr_2.abi, teph4zpr_3.abi, teph4zpr_4.abi, #%% teph4zpr_5.abi, teph4zpr_6.abi, teph4zpr_7.abi, teph4zpr_8.abi #%% [files] #%% use_git_submodule = MgO_eph_zpr #%% files_to_test = #%% teph4zpr_8.abo, tolnlines= 57, tolabs= 3.000e-02, tolrel= 6.000e-03 #%% [paral_info] #%% max_nprocs = 10 #%% [extra_info] #%% authors = M. Giantomassi #%% keywords = NC, DFPT, EPH #%% description = Convergence of ZPR wrt q-sampling with Sternheimer method #%%<END TEST_INFO>
Execute it with:
abinit teph4zpr_8.abi > teph4zpr_8.log 2> err &
To plot the spectral function A_\nk(\ww) in an easy way, use AbiPy to extract the data from the netcdf file. We can do it in two different ways:
- using a small python script that calls the AbiPy API
- using abiopen to interact with the netcdf file inside ipython
The python script is reported here:
Script to plot A_\nk(\ww)
#!/usr/bin/env python import sys from abipy.abilab import abiopen # Get file name from command line and open the file filepath = sys.argv[1] abifile = abiopen(filepath) # Plot Sigma(omega), A(omega) and QP solution # Adjust the parameters according to your system as AbiPy cannot (yet) read your mind abifile.plot_qpsolution_skb(spin=0, kpoint=[0, 0, 0], band=7)
Alternatively, one can use
abiopen.py teph4zpr_8o_DS1_SIGEPH.nc
to open the SIGEPH.nc file inside ipython. Then, inside the ipython terminal, issue:
%matplotlib abifile.plot_qpsolution_skb(spin=0, kpoint=[0, 0, 0], band=7)
to plot \Sigma_\nk(\ww) and A_\nk(\ww) at the \Gamma point for band index 7 (conduction band maximum). Note that in python indices start from 0 whereas ABINIT uses Fortran conventions with indices starting from 1. In a nutshell, one should substract 1 from the Fortran band index when calling AbiPy functions.
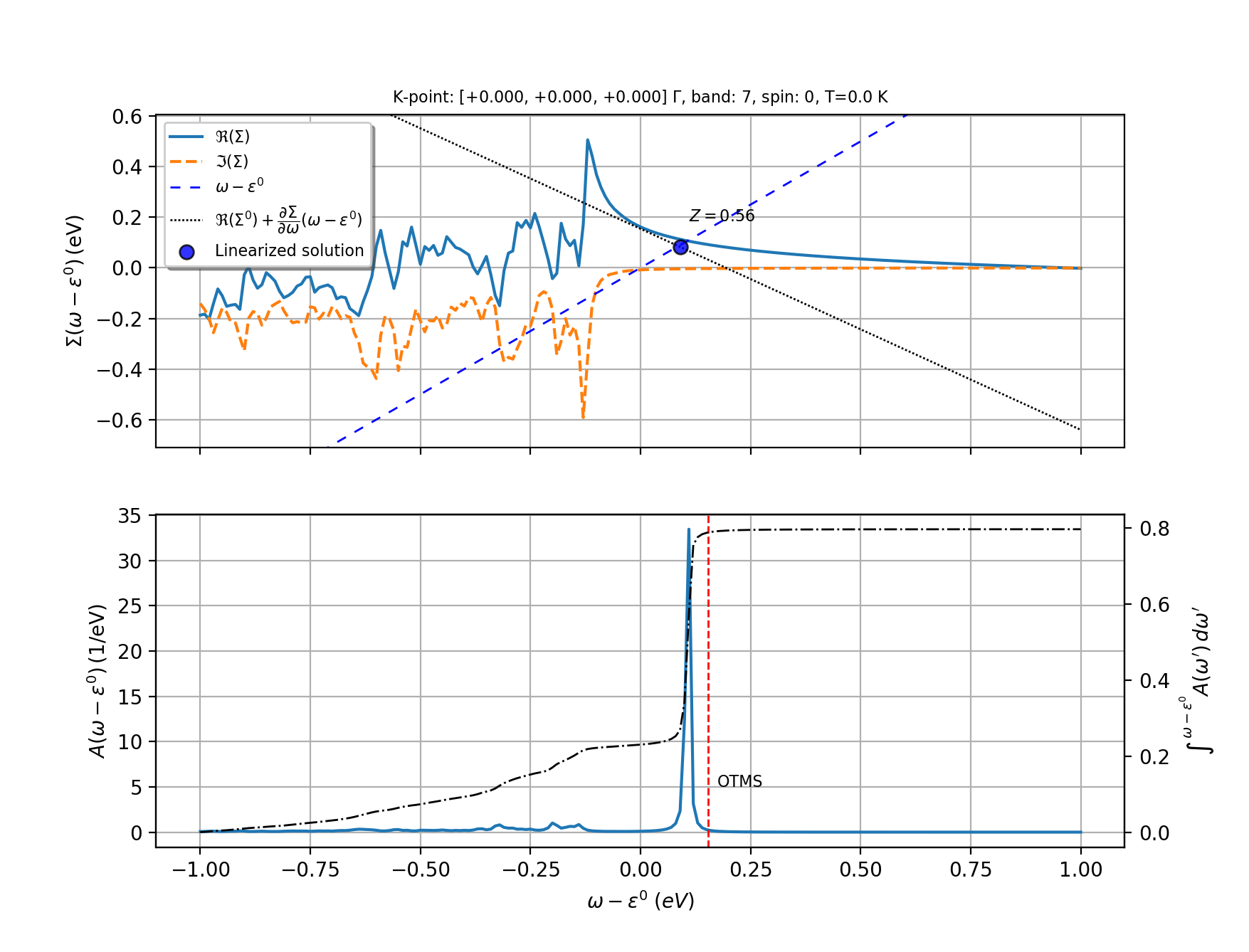
Some comments are in order here.
(i) The intersection between the blue solid and dashed lines gives the graphical solution of the non-linear QP equation:
restricted to the real axis whereas the blue circle represents the solution obtained with the linearized QP equation with the renormalization factor Z. For this particular state, the real part of \Sigma is almost linear around \ee^\KS_\nk and the linearized solution is very close to the non-linear one. There may be cases, however, in which the real part is highly non-linear and the two approaches will give different answers.
(ii) The QP solution is located inside the KS gap where \Im\Sigma is zero (well, strictly speaking, it is very small yet finite because we are using a finite zcut in the calculation). As a net result, A_\nk(\ww) presents a sharp peak in correspondence of the QP energy. Note also that our computed A_\nk(\ww) does not integrate to 1 because the number of points nfreqsp is too small and part of the weight is lost in the numerical integration. This can be easily fixed by increasing the resolution of the frequency mesh.
(iii) For this particular \nk state, \ww - \ee^0 has a single intersection with \Re \Sigma_\nk(\ww). However, it is possible to have configurations with multiple intersections that may lead to some background and/or additional satellites in A_\nk(\ww) provided the imaginary part of \Sigma_\nk(\ww) is sufficiently small in that energy range.
(iiii) The vertical red line in the second figure gives the on-shell QP energy. As already mentioned previously, the OTMS energy derives from a static formalism that reduces to the linearized QP result only if Z = 1 so it is not surprising to see that the QP peak differs from the OTMS energy. Still, this does not mean that the OTMS is wrong. Actually, it is more puzzling the fact that the background in A_\nk(\ww)is strongly suppressed and no phonon satellite is clearly visible. Obviously, our entire discussion is based on completely underconverged results but the more careful investigation reported in [Nery2018] indicates that an accurate description of dynamical effects requires the (approximate) inclusion of additional Feynmann diagrams through the cumulant expansion and that the OTMS approximation gives QP energies that are much closer to the QP peak obtained with the cumulant method.
This (lengthy) discussion was somehow needed to justify the reason why we have been focusing on the OTMS results in most of this tutorial. Now we focus on more technical aspects and, in particular, on how to compare spectral functions and self-energies obtained with different settings.
Remember that in teph4zpr_8.abi we computed A_\nk(\ww) with/without the Sternheimer method. So the question is “how can we compare the two calculations and what can we learn from this analysis?”
To compare the results obtained with/wo the Sternheimer, use abicomp
abicomp.py sigeph teph4zpr_8o_DS*_SIGEPH.nc
and then, inside ipython, type:
%matplotlib robot.plot_selfenergy_conv(spin=0, kpoint=0, band=7)
to produce the figure below with the real part, the imaginary part of the self-energy and the spectral function obtained with the different approaches:
![]()
Note the following:
(i) The imaginary part of \Sigma_\nk(\ww) is not affected by the Sternheimer method as long as we explicitly include enough nband bands around the \nk state to account for phonon absorption/emission with the full dynamical self-energy.
(ii) The real part of \Sigma_\nk(\ww) obtained with the two methods differ but the results should be interpreted with a critical eye. The Sternheimer method, indeed, is designed to accelerate the convergence of \Sigma_\nk(\ee^\KS_\nk) w.r.t. nband but cannot exactly reproduce the behaviour of \Sigma_\nk(\ww) at large frequencies since it is a static approximation. In other words, once the two approaches are properly converged one should see that the real part of the two self-energies agree around the bare KS eigenvalue and that the two curves stars to deviate at large \ww. This test is left as an additional excercise for volunteers.
For additional examples for Diamond, see this jupyter notebook
MPI parallelism and memory requirements¶
There is an important difference with respect to eph_task -4 (computation of the imaginary part) that is worth discussing in more detail. When computing the imaginary part at the KS energy for transport properties, the EPH code is able to filter both \kk- and \qq-points so that only the relevant states around the band edge(s) are stored in memory. Unfortunately, in the case of full self-energy calculations, this filtering algorithm is not possible and each MPI process needs to store all the \kk-wavevectors the IBZ to be able to compute the e-ph matrix elements connecting \kk to \kq. In other words, the wavefunctions in the IBZ are not MPI-distributed and this leads to a significant increase in the memory requirements, especially for dense meshes and/or large nband. Fortunately, the code is able to distribute bands among the MPI processes in the band communicator hence the memory required for the wavefunctions will scale as nband / np_band where np_band is the number of MPI processes in the band communicator (see eph_np_pqbks).
For ZPR calculations, the priorities are as follows:
-
Use enough np_band MPI processes to decrease the memory for the wavefunctions. Ideally, np_band should divide nband to distribute the work equally. The maximum number of MPI procs that can used for this level is nband but let’s not exaggerate. Keep some procs for the other MPI levels that are usually more efficient in terms of wall-time.
-
Once the memory for the wavefunctions reaches a reasonable level, activate the parallelism over perturbations to decrease the memory for W(\rr, \RR, 3\times\text{natom}). For better efficiency, eph_nperts should divide 3 * natom. As explained in the introduction page for the EPH code, this MPI level allows one to reduce the memory allocated for W(\rr, \RR, 3\times\text{natom}) so the number of procs in this communicator should divide 3 * natom.
-
If the memory for the wavefunctions and W is under control, you may want to activate the \qq-point parallelism to speedup the calculation. The memory requirements will not decrease, though.
-
Finally, use the \kk-point parallelism if there are enough CPUs available to boost the calculation. Obviously, this kind of parallelism makes sense if you are computing QP corrections for several \kk points and the number of np_kpt MPI processes should be adjusted accordingly. Keep in mind, however, that each \kk-point has a different computational cost so load imbalance is expected.
Important
Finally, remember that setting boxcutmin to a value smaller than 2 (e.g. 1.1) leads to a significant decrease in the memory required to store W while mixprec = 1 allows one to decrease the computational cost of the FFTs.